目的:
- 建立进化树
- 获得进化树中每个种的延续时间
- 获得某两个种最近共同祖先(MRCA)距离现在的时间
代码
1
2
3
4
5
6
7
8
9
10
| setwd("C:/Users/jlzhang/Desktop/age")
library(plantlist)
library(V.PhyloMaker)
library(phytools)
library(dispRity)
library(openxlsx)
library(phytools)
|
用中文名建进化树
查询学名
注意xlsx中保存的是中文名
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
| species <- read.xlsx("checklist.xlsx")
taxa.table(TPL(CTPL(species[,1])$SPECIES), "taxa_table.txt")
|
读取查询到的学名
1
2
3
| tab <- read.table("taxa_table.txt", sep = "/")
colnames(tab) <- c("family", "genus", "species")
tab2 <- tab[, c("species", "genus", "family")]
|
用V.PhyloMaker建树,速度较慢
1
2
3
4
| result1 <- phylo.maker(tab2, scenarios = c("S1"))
write.tree(result1[[1]], "tree1.newick")
tree <- result1[[1]]
|
获得每个种延续的时间
注意,这个与进化树收录的分类单元密切相关,一般不宜用于研究。
脚本来源 http://blog.phytools.org/2013/10/finding-edge-lengths-of-all-terminal.html
1
2
3
4
| n <- length(tree$tip.label)
ee <- setNames(tree$edge.length[sapply(1:n, function(x, y) which(y == x), y = tree$edge[, 2])], tree$tip.label)
ee <- as.data.frame(ee)
write.xlsx(ee, "terminal_branch_length.xlsx", row.names = TRUE)
|
查询两个分类单元最近共同祖先的节点名
1
2
3
| tree$node.label <- paste(1:tree$Nnode + Ntip(tree))
name_node <- getMRCA(tree, tip = c("Cleyera_japonica", "Rhododendron_simsii"))
name_node_ind <- tree$node.label[name_node - Ntip(tree)]
|
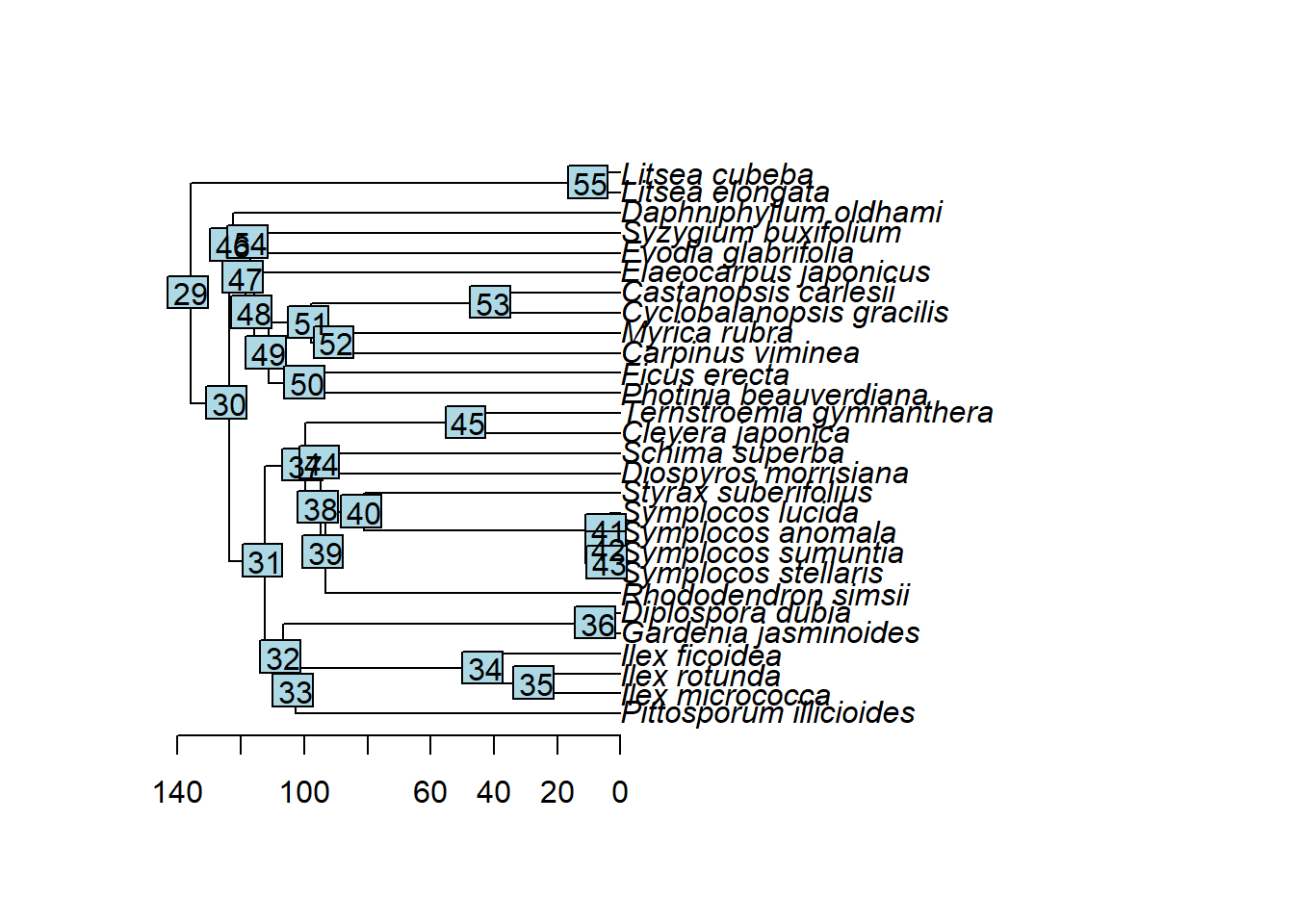
绘制进化树
1
2
3
4
| plot(tree)
axisPhylo(1, las = 1)
nodelabels(text=tree$node.label, node=1:tree$Nnode+Ntip(tree))
|
1
2
3
| plot(tree)
axisPhylo(1, las = 1)
nodelabels(paste("MRCA:", name_node), name_node, frame = "r", bg = "yellow", adj = 0)
|
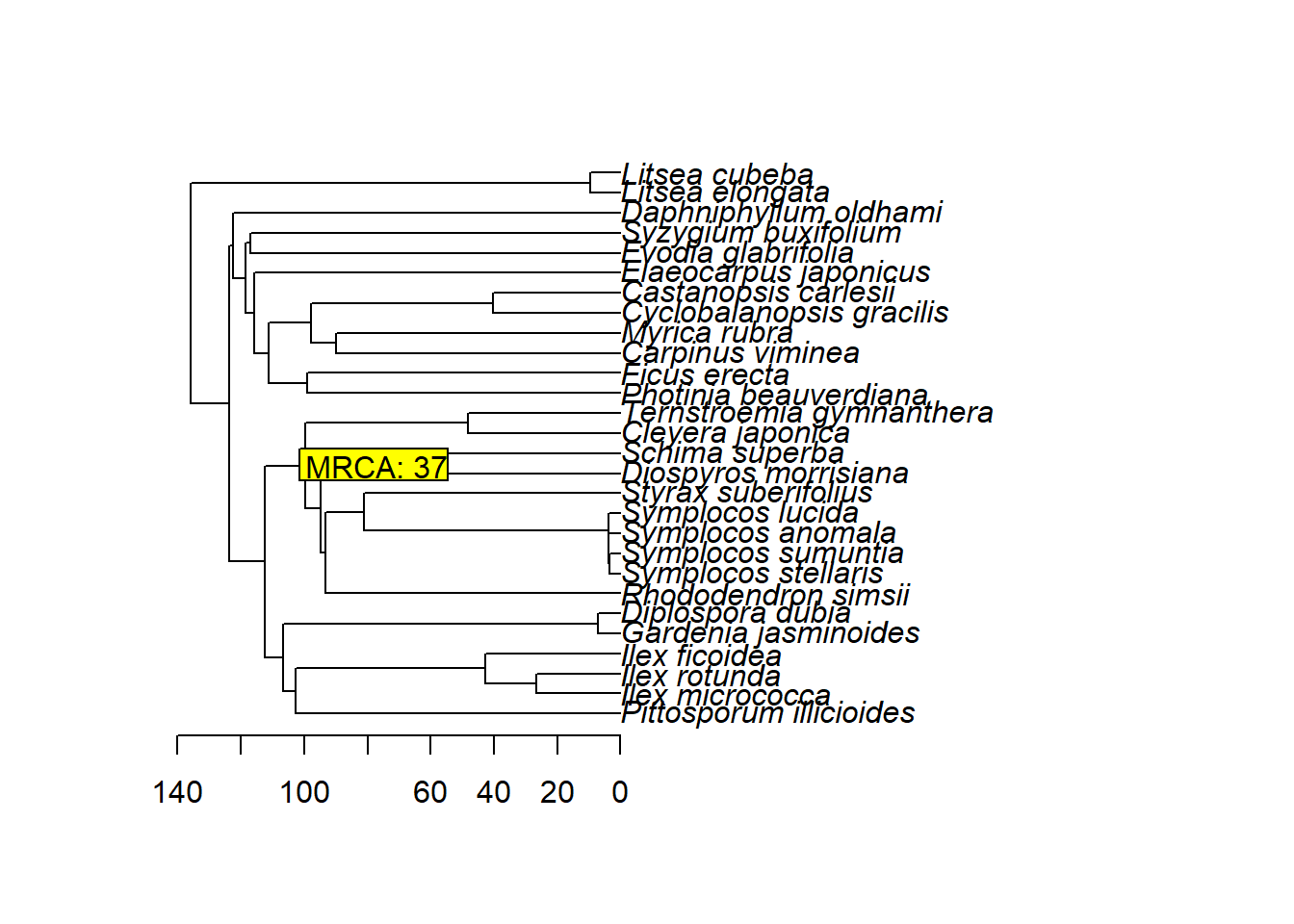
查询两个种的最近共同祖先MRCA的时间
1
2
3
4
5
|
age_dat <- tree.age(tree)
age_dat[as.character(age_dat$elements) == name_node_ind, ]
|
更多内容