系统发育独立差(Phylogenetic Independent Contrast, PIC)是去除性状分析中物种系统发育关系的一种方法,是美国进化生物学家 J. Felsenstein于1985年提出的。
当时,有动物学家检验了若干哺乳动物脑容量和体重的相关性。然而,所用统计方法都假设各个种的性状是相互独立的,采集自同一个正态分布的总体。由于不同种之间存在系统发育上的联系,因此不能直接套用常规统计模型,所以所得结果也就不可信了。在Felsenstein之前,一些学者已经意识到这个问题,但是都没能给出理想的解决方案。
Felsenstain认真思考了这个问题,并且认为:研究多个不同性状的相关性时,必须考虑系统发育关系。他提出:假设物种性状的进化服从布朗运动,那么用系统发育关系上最近的分类单元性状的差值,再经过枝长加权,就可以得到一组新的数据。这组新的数据去掉了进化信息的干扰,因此可以用常规统计方法检验。上述方法新获得的数据就称为系统发育独立差 (The Phylogenetic Independent Contrast, PIC)。

图1. Felsenstein (1985) 的论文
举例来说,如果要研究哺乳动物脑容量和体重的关系,就要先获得这些种之间的进化关系,也就是经过分子钟校订的进化树。然后再计算脑容量的PIC和体重的PIC,再用脑容量的PIC和体重的PIC建立线性模型,进行相关性检验。
如果假设每个性状的进化服从布朗运动模型,也就是每个子代的性状的值是从祖先节点的性状随机变化而来的,那么进化树上相邻节点的两个种性状偏离于最近共同祖先性状的期望应该是0,且二者之间的差异正比于分化时间,也就是进化树的枝长。
在1985年的论文中,Felsenstein提出了如下算法:
- 假设相邻的分类单元为i和j,其性状分别为X_i和X_j,i和j的共同祖先是节点为k。
- 先计算X_i - X_j,作为X和Y的contrast。该contrast的期望为0,方差与枝长v_i+v_j成正比
- 从进化树中去掉节点i和j,保留节点k,将k当做末端节点。具体来说,就是以X_i和X_j的加权平均值作为X_k,公式如下:
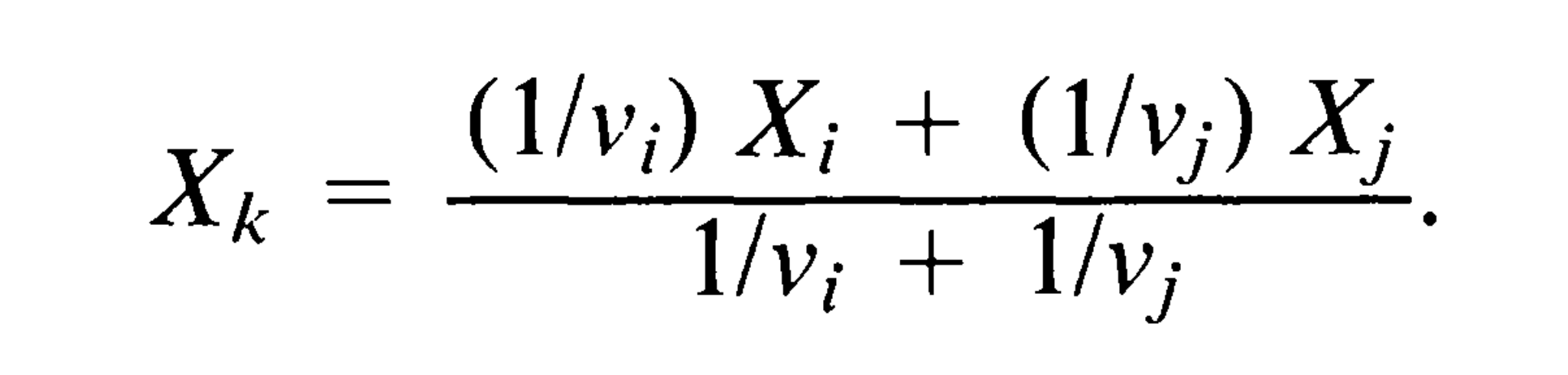
- 将k节点下的枝长从v_k延长为v_k + v_i**v_j*/(v_i + v_j)。这样做的原因是,第3步中,k节点的X_k并非真实值,而是用X_i 、 X_j计算到的。
经过1-4步,进化树上的末端分类单元就减少了1个,同时,也计算出了一个contrast的值。
重复1-4步,直到进化树中只有一个节点。如果进化树中有n个末端节点,那么上述过程将得到*n-*1个独立差(contrast)。图2-3展示了只有五个物种的进化树,PIC的计算公式。
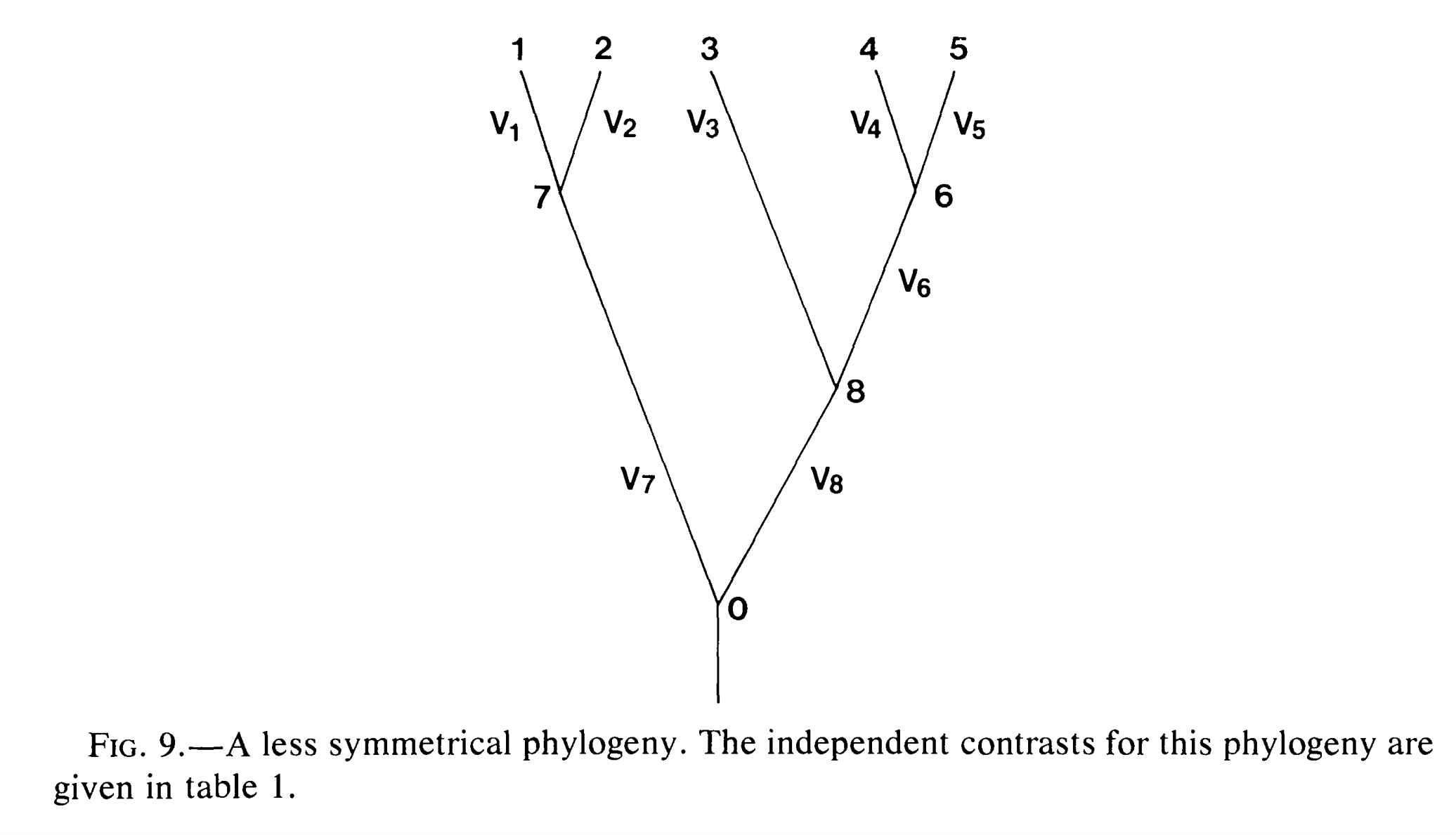
图 2. 拥有5个分类单元的进化树,分类单元名称(1-5为末端节点,6-8为内部节点)、枝长(V)

图3. PIC的计算公式
ape程序包提供了计算PIC的函数pic(),该函数使用的正是Felsenstein (1985)的算法。一般用户并不需要了解函数内部的机制,只需要提供进化树和性状向量即可。
以下为ape程序包中pic函数的示例:
1 | library(ape) |
从以上结果可以看出,将性状直接进行相关分析以及用性状的PIC进行相关分析,所得结果是有差异的:去除了系统发育关系之后,性状X和性状Y之间的相关性降低了。去除系统发育关系之前 (Pearson’s r = 0.8296, t = 2.5736, df = 3, p-value = 0.08224),去除系统发育关系之后(Pearson’s r = -0.5179156, t = -0.85623, df = 2, p-value = 0.4821 )。
这里比较难以理解的是,计算出来的PIC总是比真实性状数据少一个,但是不同性状的PIC都是比真实数据少一个,用不同性状的PIC向量建立线性模型,所得结果,就是我们要计算的性状之间的相关性。
ape的pic函数要求不同种性状出现的顺序与在进化树中出现的顺序完全相同。为此,picante程序包提供了match.phylo.data()函数,只要用户为性状数据(无论是data.frame还是vector)提供了对应的物种名(如果是多个性状,格式应该为dataframe,其行名应该是物种名),该函数就能将性状数据按照进化树中出现的顺序排好,非常方便。
以下代码展示的就是怎样通过植物物种名(拉丁名)获得进化树,再计算植株高度的PIC,源数据来自S. Kembel博士的培训资料。
Felsenstein在1985年的论文中说,他提出的算法可计算多分枝结构的PIC,但是ape的pic函数不能处理含有多分枝结构的进化树,所以下面的代码调用了multi2di()函数,将进化树转换为二叉树。转换的方法是为每一个多分支节点随机添加一个很小的枝长,由于枝长很小,对结果的影响很小,几乎可以忽略不计。
1 | setwd("...") |
参考资料
- Felsenstein, J. (1985) Phylogenies and the comparative method. American Naturalist, 125, 1–15.
- Jin, Y., & Qian, H. (2019). V. PhyloMaker: an R package that can generate very large phylogenies for vascular plants. Ecography, 42(8), 1353-1359.
- Kembel, S. W., Cowan, P. D., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., … & Webb, C. O. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics, 26(11), 1463-1464.
- Kembel S. 编, 张金龙 译, 在R中分析物种和系统发育多样性 http://blog.sciencenet.cn/home.php?mod=space&uid=255662&do=blog&id=1116212
- Paradis, E., & Schliep, K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics, 35(3), 526-528.
- Zhang, J.L. (2018). plantlist: Looking Up the Status of Plant Scientific Names based on The Plant List Database. R package version 0.6.1. https://github.com/helixcn/plantlist/