PDF:群落系统发育常见问题合集 张金龙.pdf
R Markdown 源代码 https://github.com/helixcn/comphylo_faq
入门 群落系统发育入门资料 提问 :
能不能介绍些phylogenetic方面的入门资料及其在生态学方面的应用呢?
回复 :
系统发育分析入门最好的教材,我觉得是 The phylogenetic handbook。
另外,我觉得先学习几个软件的基本操作流程,并且从建树的原理结合学起从CLUSTAL, MUSCLE、Phylip,PAUP*, MrBayes, RAxML, BEAST, r8s等各种软件,都需要认真阅读使用手册。 Bodega Phylo Wiki也是一个理想的去处。
Phylogenetic structure等内容的理解,还需要结合文献,同时认真学习Phylocom或者picante等软件的说明书。
我的网页上也链接了一些资源,http://jinlongzhang.weebly.com/resources.html 这些幻灯片和软件操作指南都是可以下载的。
怎样学习系统发育软件? 提问 :
对于怎么入手去学习谱系软件,你有什么好的建议吗?
回复 :
学习系统发育分析,一定要先把R软件学好,特别是ape程序包和picante程序包,弄清楚随机化检验的原理及实现方法,同时要学习Phylocom软件。 只要按步骤操作就可以了。但是这些只是技术手段,都是比较容易实现的。真正决定研究水平的,是研究所解决的问题,如检验假说,这就需要阅读大量的文献。
建议学习Nathan Swenson, Steve Kembel, Camb Webb, Mac Cadotte等人的论文,从理论上进一步提高。
进化树枝长的意义 提问 :
http://evolution.berkeley.edu/evolibrary/article/phylogenetics_02 中说系统发育分析中,节点表示共同祖先,枝长表示改变了的性状的数量(进化速率?)?
那么,如果要比较两个物种的出现早晚,就比较它们到共同祖先的枝长就可以了?
如果其中一个物种到共同祖先之间的节点数比另一个物种多,只能说明在它这条分枝上保存下来的物种比较多,而不能说明它这条分枝的进化速率比较快?
回复 :
物种到共同祖先的时间是一致的。而不同类型的进化树, 其枝长的意义是不同的。
Phylogram中有一类, 是Chronogram, Chronogram的枝长就是表示时间, 经过分子钟校订之后的进化树, 都是Chronogram。Phylogram还包括, 简约法获得的进化树, 这里的枝长, 表示变化了的为点数。极大似然法获得的进化树中,枝长表示碱基突变的概率。所以不能光看枝长, 首先要了解是怎样获得的进化树。
为什么NRI/NTI的临界值是2, 而Pwd和Dnn的临界值是0? 提问 :
近日研究进化多样性,看到一篇文献Stochastic and deterministic assembly processes in subsurface microbial communities。文中讲到NRI临界值的判断方法,for single community,临界值是2;across all communities,临界值是0。实在想不通single community和all communities的区别,为什么single community下临界值是2,为什么all communities下是0.
我的理解是:
single community时候,null model随机抽样的样本不等于总体;样本不等于总体的时候抽样结果需要进行参数估计,此时抽样结果可以表示为 mean±t*sd。所以如果想在95%的confidence下,对结果进行判断,此时t对应正态分布界值表中的1.96。all communities的时候,null model随机抽样的样本等于总体;不需要进行参数估计。我的理解很牵强,是从数字上推断的,手头也没有找到关于null model更详细的资料。
回复 :
我觉得您的理解是正确的, 一般认为NRI或者NTI大于1.96或者小于-1.96的结果, 在95%的水平是显著的。 这是因为计算NRI或者NTI都是Standard effective Size的变型。 两者都是基于多次随机化的群落或者进化树计算的。 随机样本量很大的情况下, MPD或者MNTD的分布会接近正态分布。 所以文中作者所言接近于2, 应该是接近1.96, 才是正确的。 across all communities,临界值是0,可能是指系统发育距离距离或者发散的趋势, 与上文的NRI或者NTI是否显著为不同的东西。
NRI和NTI与0的关系 提问 :
我计算了NRI和NTI指数,是否可以以0作为标准:大于0,谱系聚集;小于0,谱系发散?
回复 :
可以这么说的, 以正负作为标准, 只能说有聚集或者发散的趋势,但不能提供显著水平。
Phylomatic 用Phylomatic建树的时候,需要检查植物是否是异名吗? 提问 :
最近读了你的科学网安装了最新版本plantlist包,生成科/属/种列表,并用status函数逐一查验学名是否接受,不接受者重新查其学名更正直至status状态为accepted 或者synonym(synonym也是接受的意思对吗?)。做完这些工作后用Phylocom的bladj模块为进化树拟合枝长,问题出现在每次生成的out.tre文件无法打开,显示“Cannot open phylogeny file”,用Figtree也打不开。是因为我的物种学名依旧有问题,还是其它原因呢?我也选取部分物种来重复同样步骤却也一样的out.tre文件无法打开。
回复 :
请参考下面的意见。
现在通过Phylomatic可以直接生成有枝长的进化树, 无需再用BLADJ。我帮你建好了进化树,但是有两个分类单元没有找到:
1 [NOTE: 2 taxa not matched: Pinaceae/Abies/Abies_chensiensis, Pinaceae/Larix/Larix_principis-rupprechtii, ];
有几点需要留意:
物种名在数据分析阶段不要包括命名人
synonym是异名, 是分类学上不获接受的名称。 不过这是The Plant List网站给出的意见, 只代表某种观点, 不能认为《北京植物志》或者《河北植物志》等地方植物志给出的结果是错误的。 当然, 对于拼写有错误的学名, 是必须要检查出来并改正的。 status的结果只表示该学名是否接受,以便检查拼写。
在获得taxa_table.txt的科属种名单之后, 用Phylomatic建立进化树时, 应该选用Zanne 2014的进化树作为骨架
应该尽量用Phylomatic生成有枝长的树 提问 :
现在正在作种子大小进化这一块的研究工作,要用到Phylocom这个软件,但是在使用BLADJ计算进化枝长时,系统总是提示”“0x00423382”指令引用的”0x0136812”内存,该内存不能为read”,不知道问题出在哪里,请给予帮助,万分感谢!!!
回复 :
您遇到的情况是Phylocom自身的问题,目前也没有很好的解决方法。如果在进化树中有非ASCII字符,以及枝长中出现科学计数法,Phylocom会自动停止工作。当然,还存在很多意想不到的情况。建议不要再使用BLADJ以及WIKSTROEM的分化时间,而是用Phylomatic直接在线生成有枝长的进化树。
方法如下:
打开 http://phylodiversity.net/Phylomatic/
将 科属种列表 拷贝到 taxa = 框中
storedtree = 选择 Zanne 2014
勾选 clean = TRUE, 其余选择默认.
点击Sent 按钮
稍等片刻, 将网页中的输出结果拷贝到文本文件中, 并另存为 phylo文件。注意, 该phylo文件不能有任何扩展名。该进化树为Newick格式,不含Singletons, 因此可以用R的ape读取或者用Figtree软件绘图。
我在博客中也进行了相应的更新 http://blog.sciencenet.cn/blog-255662-335503.html
怎样获得Ultrametric树? 提问 :
对 Ultrametric tree插入类群的话,除了用Phylocom还有什么其它方法吗。这棵 Ultrametric tree我们已经获得了相应的时间树,已知年龄的节点很多,在用BLDJ前准备的age文件我觉得不太好准备,还是有什么简单的方法?另外,我不确定这样是不是改变了原来已有类群的枝长(Phylocom我还不是很熟,对这点有疑问),而我们想保持树上原来有的类群枝长不变。非常感谢~
回复 :
这种情况我觉得直接编辑进化树比较好, 可以参考 http://treegraph.bioinfweb.info/Help/wiki/Inserting_nodes
Phylomatic适于基于科属种名录以及现有的backbone获得进化树。 在2011年时, 我用Webb提供的Davies科级进化树作为骨架, 获得过种等级的进化树。 Phylomatic有本地版本的, 如果将已经获得的属级进化树内部做上足够的标记, 相信它是可以帮忙做出进化树来的。
建树时物种名不要含有命名人 提问 :
正在学习您的博文“用Phylomatic和Phylocom进行群落系统进化分析(20160105)”,遇到两个问题,请教一下:
对数据phylo.txt,在使用Phylomatic建树,网页出现下面提示:
1 Server error!The server encountered an internal error and was unable to complete your request.Error message: End of script output before headers: pmwsIf you think this is a server error, please contact the webmaster.
2.对数据res.sp.smt.txt,读树时,出现下面错误:
1 > tree.jgs <- read.tree("phylo.txt")Error in read.tree("phylo.txt") : The tree has apparently singleton node(s): cannot read tree file.Reading Newick file aborted at tree no. 1
回复 :
您的进化树中物种名含有很多空格,而且物种名还包含命名人, 这在R读取中会遇到困难。 在提交物种名单到Phylomatic时,就应该保证学名只包括英文字母以及“_”, 不要包含任何其他的字符,否则即便生成了进化树, 在R或者其他进化树处理软件中,后续数据分析会很困难。 特别是命名人中还有英文括号, 例如 Padus_racemosa_(Lam._)_Gilib., Swida_macrophylla_(Wall.)_Soj¨¢k 等, 会影响进化树的读取。英文括号在newick文件中是用来定义进化树拓扑结构相关的,所以物种名中不要包含括号。
为什么要用Zanne2014进化树骨架? 提问 :
我最近正在学习您的 “用Phylomatic和Phylocom进行群落系统进化分析(20160105)”
枝长(即所谓的年龄长吗?)是一个相对值吗(因为我发现同一物种在不同物种组合中结果不同)?
回复 :
Zanne 2014是目前基于分子序列的最完整的进化树, 所包含的科属种也是最多的,选择该进化树拟合枝长,结果会更可靠一些。
为什么用Phylomatic建树要选择 CLEAN? 提问 :
请问为何要这样设置Phylomatic “storedtree = 选择 Zanne 2014;clean = TRUE, 其余选择默认.”最后输出的结果,数值代表什么?
回复 :
选择clean = TRUE, 是因为Phylomatic在数据处理时, 如果选择默认, 会在某一个科下面, 多增加很多层括号, 而这种多层次的括号, 在newick格式的进化树中是不允许的, 因为newick格式规定, 每一个分类单元分化成两个或者多个子代时, 才增加一层括号, 如果没有分化, 则不应该增加一层括号.Phylomatic增加很多层括号, 是为了Phylomatic拟合时间方便, 但是这样就不可避免了引入了很多孤立的分类单元. 这种分类单元称为singletons, 在APE以及Figtree等软件都不能处理. 所以为了能让进化树在R中处理, 还是要选择clean,去掉那些多余的括号, 以免singletons出现.
Phylomatic生成进化树的枝长 提问 :
Phylomatic生成进化树的枝长是什么意思?
回复 :
输出的枝长, 就是该分类单元的持续时间, 单位是百万年.同一个种的年龄应该是一致的, 当然, 受到物种名录的影响, 进化树的拓扑结构可能不同, 这就造成种的年龄在不同的物种名录中出现不同的结果.
为什么用Phylomatic建树, 有些属名会消失? 提问 :
我在用Phylomatic建属级水平的树(taxa里格式:family\genus)用zanne2014与R20120829都能成功,但是发现有些属丢失了(Ficus、Alnus等,taxa里是有的),为什么出现这种情况?按理来说匹配不上应该会报错。是不是我的建树方法不对,taxa里必须是:family\genus\species的格式?如何获得属级水平的树(树的tip为属)?
回复 :
属一级的进化树, 其实是每一个属选择一个种, 而不建议科属种名单只到属的水平。如果用科属种的名单再有一些种丢失,仍然应该是Phylomatic内部的bug。 相关的bug可以告诉Cam Webb, 网址是 https://github.com/camwebb
为什么用Phylomatic建树,有些科的植物会找不到? 提问 :
我看您输出的进化树后面有7个taxa无法匹配,是否是因为APG Ⅲ里面还没将这些科分开(当然里面有5个是棕榈科,是否APG Ⅲ不含单子叶的?)还是科名与APG Ⅲ里的不匹配,虽然这些科名是从The Plant List获取的?之前听植物所刘冰博士介绍APG Ⅲ已经将科之间的关系已经区分清楚了,我比较纳闷怎么还有双子叶科(例如其中的五桠果科)不能匹配?
回复 :
棕榈科的名称有两个Palmae 和 Arecaceae
1 Palmae= ArecaceaeGramineae= PoaceaeLeguminosae= FabaceaeGuttiferae= ClusiaceaeCruciferae= BrassicaceaeLabiatae= LamiaceaeCompositae= AsteraceaeUmbelliferae= Apiaceae
我在校对plantlist程序包时, 已经将Palmae改为 Arecaceae, 但是如果Phylomatic网站所存储的Zanne2014进化树中用的是Palmae这个科名, 就会出现不匹配的情况。此时可以尝试更改科名为 Palmae。
若Zanne2014的进化树中, 不包含Dilleniaceae, 或者Dilleniaceae的名称拼写错误, 也会出现不匹配的情况. 因为涉及到网站编程, 如awk语言等, 我也不是很清楚。
Phylomatic只用科名可以建立进化树吗? 提问 :
我最近正在使用 Phylocom和Phylomatic进行群落系统进化分析,在进行分析的过程中出现一个问题,举例说明如下:物种名录(已通过R package (plantlist)进行科属的查询):
1 Salicaceae/Ahernia/Ahernia_glandulosaBegoniaceae/Begonia/Begonia_cathayanaPentaphylacaceae/Adinandra/Adinandra_epunctataMyrtaceae/Decaspermum/Decaspermum_gracilentum
然后只将科名放入Phylomatic建树得到结果如下:
1 ((((((((((((((((((((((((((begoniaceae)))))cucurbitales))),((((((salicaceae))))malpighiales))celastrales_to_malpighiales))fabids))rosids),(((((((((pentaphylacaceae))))ericales)ericales_to_asterales)asterids)))))core_eudicots)trochodendrales_to_asterales)sabiales_to_asterales)eudicots)ceratophyllales_and_eudicots)poales_to_asterales)magnoliales_to_asterales)austrobaileyales_to_asterales)nymphaeales_to_asterales)angiosperms)seedplants)euphyllophyte;
再使用Phylocom软件用bladj为进化树拟合枝长,得到进化树如下:
(图从略)
结果仅显示了三个科的进化树,还有一个科(Myrtaceae)在树种并未显示。我所分析的全部数据总共有187个科,其中有12科为在进化树中显示。请问出现这样的结果是我的操作上出现了问题,还是Phylomatic系统中就没有这些未显示科的信息。我应该怎么处理这个问题?
回复 :
Phylomatic是用来建立种一级的进化树的, 不能用来建立科等级的进化树。 必须将 Salicaceae/Ahernia/Ahernia_glandulosa 这个文件提交到Phylomatic。
Phylomatic进化树的枝长可以到种吗?网上有带枝长的进化树吗? 提问 :
Phlocom中的进化枝长可以到种么?还是只能到属,还是只有部分能到属?
网上能找到带枝长的megatree或其newick文件么?只有某个科的也可以,比如菊科、禾本科和豆科的。
回复 :
Phylocom的进化枝长只是精确到科,每个种的分化时间在科以下三等分。为此我做了一个图, 表示在种水平以及属水平,Phylomatic和Phylocom的BLADJ是如何处理的。
网上是可以找到带枝长的进化树的, 例如 https://github.com/camwebb/Phylomatic-ws/tree/master/data
种以下分类单元,如亚种和变种,在Phylomatic中应该如何处理? 提问 :
种以下分类单元,如亚种和变种,在Phylomatic中应该如何处理?
回复 :
最后一个问题, 亚种和变种,可以考虑合并为一个种, 也可以考虑作为一个独立的分类单元对待。 但是在材料与方法这部分一定要明确说明。说清楚的情况下,审稿人一般不会太刁难。
Phylomatic能用于动物系统发育结构分析吗? 提问 :
你上次介绍的Phylomatic是致力于解决植物系统的进化问题,似乎没有涉及到动物系统。我想问下你如果要根据现有的形态或者说是分类系统建一个无枝长的系统树是不是只能有手输入的比较笨的办法?是否还有什么比较好的办法?
还有Phylocom主要是利用bladj模块对建好的树进行一个赋值,建一个有值的树,我想问的是那个模块是不是就是指那个wikstrom.ages文件,利用那个文件中的数据对我们的树进行赋值,还是如果我们需要进行对动物的系统树的建立必须自己先建立一个赋值文件,然后进行树的赋值?如果是那样的话,那个赋值文件时怎么设置的呢??
回复 :
Phylomatic是权宜之计,因为植物测序的还比较少。正常情况下是应该用DNA序列建立进化树的。Phylocom程序包中有一个单机版的Phylomatic,似乎提供相应的进化树框架就可以了。
动物的进化树框架,我目前也不清楚用什么版本,但是用分子的方法是肯定的。相信国际上已经有相应的各主要动物类群的进化树,可以搜索一下看看。因为我自己不研究动物,真的没有很好的解决办法。
我想最好的方案,就是下载各序列,比对好后拼接在一起。之后,用MrBayes或BEAST者PHYML或者RAxML建立进化树,再对进化树进行校对,如NPRS改正等。这样的出来的进化树,用在任何分析,也不会有人提出过多的质疑了。
您提高的时间赋值文件, 请参考wikstrom.ages文件的格式,但是实现起来应该不是特别简单, 涉及到很多编辑工作,具体建议问一下Cam Webb。
Phylomatic的进化树能包括蕨类植物和裸子植物吗? 提问 :
文献只提及用Phylocom的bladj结合wikstrom的分化时间建树,2001年的文章,我也看了您所做报告PPT中建树方法的介绍,但是我还是没有搞清楚,想请教您一下:
建树前应准备的数据(是不是需要节点的顺序和节点的进化时间),用什么算法,在什么软件中进行?我后续的工作想进行phylogenetic independent contrast、 partial mantel tests 和PCA分析,对生成的树有什么要求?
Phylomatic中不能匹配的物种和裸子、蕨类植物怎么一同建立在进化树中?
回复 :
Phylocom的BLADJ校订时间, 用的是Wikstrom的年龄, 这个文件在Phylocom软件的发行版中是有的。
PIC对进化树没有过多的要求, 如果用Phylocom进行PIC, 则执行的是Garland等人提出的方法, 对结果进行了一定的修正。Partial mantel test需要的是距离矩阵, 记得用vegan或者 ade4 程序包, 可以做。
Phylomatic提供的进化树的backbone,是有花植物的, 对于其他植物没有很好的办法,除非提供有效的Backbone,否则是难以整合的。此外, 由于蕨类等和被子植物很早就区分开了, 它们的枝长很长, 对结果的影响可能会很大, 这是需要注意的。
科属种等级的进化树应该怎样用Phylomatic建立?枝长应该怎样提取? 提问 :
Phylomatic要求的输入格式是family/genus/species,但是如果只输入family也能生成Newick格式的进化树,这两个有什么不一样啊?
有些植物虽然是被子植物为什么不能匹配,最后输出的Newick中没有该物种(family/genus/species输入)或者该科(仅family输入)?
我现在提取枝长是通过修改txt文件中的格式,再导入excel得到枝长,在R中提取枝长的函数是什么?
回复 :
Phylomatic要求的输入格式是family/genus/species,但是如果只输入family也能生成Newick格式的进化树,这两个有什么不一样啊?
Phylomatic要求输入family/genus/species格式的名录,生成的是物种等级的进化树。若只提供family,也能生成进化树,但可能只是科节点的进化树。
有些植物虽然是被子植物为什么不能匹配,最后输出的Newick中没有该物种(family/genus/species输入)或者该科(仅family输入)?
被子植物的科名,有若干通用名,包括:
1 Palmae= ArecaceaeGramineae= PoaceaeLeguminosae= FabaceaeGuttiferae= ClusiaceaeCruciferae= BrassicaceaeLabiatae= LamiaceaeCompositae= AsteraceaeUmbelliferae= Apiaceae
若所属物种是上述几个科的, 可能存在名称不匹配的情况, 则请换成Phylomatic能识别的科名。部分科,如葡萄科,目前的版本,由于Zanne2014进化树的限制,还不能识别。暂时没有更好的办法。
我现在提取枝长是通过修改txt文件中的格式,再导入excel得到枝长,在R中提取枝长的函数是什么?
如果是用read.tree生成的phylo文件, 则枝长为phylo文件的edge。
为什么有些物种Phylomatic不能识别? 提问 :
根据您的文章《用Phylomatic 和Phylocom进行群落系统进化分析》提供的方法做了一个系统进化树,但是在做的过程中,发现有一个物种(Allium polyrhizum )无法识别,这种情况应该怎么处理呢?
回复 :
Phylomatic 软件在建立进化树的时候,识别物种主要是看其所在的科Family的,因此提供一个包含科属种的taxa table就可以了。 taxa table是 family/genus/species的格式,用向右的斜线隔开。Allium polyrhizum 这个种,按照APGIII系统应该放在 Amaryllidaceae (参见http://www.theplantlist.org/browse/A/Amaryllidaceae/ ), 该网站可以查询某一个属所在的科, 按照要求填写好taxa table即可。
Phylomatic是否能建立只包含科名的进化树? 提问 :
我只需要建立科级水平的系统发育树,那我是否可以只在Phylomatic里输入科名(APG科/我的植物科)?还是我应该每个科选取一个代表种,然后用科/属/种的格式输入Phylomatic?
我的数据是51个科的植物,但是建树时我发现输出的系统发育树中只有50个科,禾本科 (Poaceae)不在树中,请问这是什么原因?我应该如何解决呢?
回复 :
如果用Phylomatic建立进化树, 物种名是什么都可以,因为它只是一个标签。 建议是每个科选取一个代表种, 然后按照 科/属/种 的方式建树即可。
我不太清楚你的建树的物种列表是怎样的, 所以不好做出推断。 所有的科名,都应该是 apgiii系统的接受名, 而 Poaceae是没有问题的。
为什么Phylomatic软件中有些植物的科名不匹配? 提问 :
我将名录用在线Phylomatic计算后,返回的结果中有些物种not matched。这些植物都是经过你的plantlist软件包检测的,我用在线工具plantminer也是可以查询到并显示Accepted的。我发现这些 not matched物种全部是葡萄科植物。另外,我在您微博中的读者留言区也看到有人也遇到这种问题,其中提到的崖爬藤也属于葡萄科。难道葡萄科都有问题?不知道为什么会这样,想听听您的建议。
匹配不了的物种名如下:
1 Parthenocissus dalzieliiVitis pseudoreticulataAmpelopsis glandulosaCayratia japonica
回复 :
Phylomatic并不检测物种名是否为接受命, 它只根据科名(部分属也许参考属名,请参考最新的Phylomatic文档)来建立进化树。我之前也遇到葡萄科植物不能匹配的情况,这应该是Phylocom网站本身的问题。具体我觉得还是要咨询Cam Webb博士。 好像棕榈科也是不能匹配的。
Phylomatic能够为哪些种建立进化树? 提问 :
我还有几个弱弱的问题想请教一下:
第一,既然Phylocom软件只能构建被子植物的谱系树,那么像冷杉群落,建群种冷杉是裸子植物,如果不计算它与其他物种之间的亲缘关系,那么做这个有何意义?
第二,为什么不删除裸子植物,在Phylomatic软件中也可以生成谱系树?而且Phylocom软件也可以生成带枝长的谱系树?这是怎么回事?
第三,在sample_data下的sample文件中的各列代表的含义具体指什么?第一列是样方名称,那么样方指的是11、5 5、2020等样方么?第二列是个体数,指的是前面1 1等样方中的所有个体数么?第三列是物种数,那么sp1是指样方1中的一个物种么?要是有很多个物种呢?比如clump2a,2,sp10具体指什么?
回复 :
第一个问题:能否计算冷杉与其他物种的亲缘关系, 取决于是否有数据。 也就是冷杉和群落内其他植物的分化时间。冷杉群落,冷杉当然是必须要考虑的。
第二个问题: 最初,Phylomatic不能生成有枝长的进化树,只能生成无枝长的拓扑结构,然后用Phylocom 的 BLADJ拟合枝长。后来Phylomatic进行了升级,如果Backbone 进化树中, 有裸子植物, 那么就能用Phylomatic生成系统树。
第三个问题: 请参阅 Phylocom的说明书。
第一列为样方名称 clump2a
第二列为个体数,也就是有2个个体
第三列为物种名 sp10
一个样方中有很多个物种,则样方名重复, 对应的物种名增加即可。 也就是用不同的行表示。
Phylomatic建的进化树有什么问题? 提问 :
之前,我用Zanne 2014版本的数据生成了带有枝长的谱系树,但是文章投出去之后,有个审稿人觉得这个树错误很多,建议我换一种谱系树重建,于是我改用原来的R20120829重建,但是将准备好的物种名录输入网页版,生成不带枝长的谱系树时,这个谱系树的结尾没有seedplants,于是后续在cmd > cd C:\Phylocom运行中也有问题。请问这个问题在哪呢?
回复 :
如果进化树中所涉及的分类单元不太多(3000之内), 而且大部分都有DNA条形码, 那么请尽量建立分子系统树 (参见 http://blog.sciencenet.cn/blog-255662-306430.html )。
Zanne的进化树是目前所知包含植物物种数最多的分子系统树。其中当然可能会有不少错误, 但是哪里错了, 最好能指出来。错误的序列, 无论是由于物种鉴定造成的错误,还是由于后续的数据处理等造成的错误, 都是对最终的系统树的拓扑结构有影响的, 但是究竟影响有多少,有很多不确定性, 是很难回答的问题。 这主要是由于建立进化树的软件,都是基于数值方法计算似然函数的最小值的。
越是年代久远的进化树, 准确性就越低, 信息缺失也是越多。R20120829 这个系统树 (参见 https://github.com/camwebb/tree-of-trees/blob/master/megatrees/R20120829.new)只有植物科水平的系统发育拓扑关系, 没有枝长, 其内部节点的分化时间需要用Phylocom的BLADJ算法拟合。 这样一来,科以下的系统发育关系就没有了。损失的信息更多,结果可能更不准确。从这点来看, 审稿人可能更不满意。
Markus Gastauer整理了R20120829这棵进化树 ,为其添加了一些内部节点的名称, 以便在Phylomatic的时候能够更好地控制时间 (https://www.ncbi.nlm.nih.gov/pubmed/27097100)。Gastauer标注后的进化树,Cam Webb已经在他的github页面公布了 (https://github.com/camwebb/tree-of-trees/blob/master/megatrees/R20160415.new)。 不过新的进化树仍然没有seedplants这个节点。所以如果真的要使用R20120829 ,不妨试试R20160415.new这个改进版本, 然后再手工编辑一下, 添加seedplants节点, 再用BLADJ拟合时间。
Phylomatic对物种数有什么限制? 提问 :
我现在想请教一下,附件是XX样地的物种名录,我copy 到http://phylodiversity.net/Phylomatic/ 以后,总是不能返回正常结果(The gateway did not receive a timely response from the upstream server or application.)。经过检查和比较,我发现当拷进去360个左右的物种名时,就能正常返回结果。而总的物种名是390,遇到这种情况应该怎么处理?能不能先生产两个tree再进行合并?望指教。
同时,我看见你在1月5日对你博客中对Phylomatic建树方法进行了更新,在文章中还需要特别指出么?谢谢
回复 :
我将物种名单拷贝到Phylomatic上, 也出现了你所述的问题,我觉得可能是服务器有一些限制。可以考虑用R的brranching包(https://cran.r-project.org/package=brranching),或者用S.Phylomaker(https://github.com/jinyizju/S.PhyloMaker)试试。
Phylomatic建树分类单元数太多怎么办? 提问 :
我目前针对中国常绿阔叶木本植物进行研究,物种数有6266种,想尝试建立这些物种的系统发育树。但是:
在使用Phylomatic时,由于物种数太多,Phylomatic显示“Trees/taxa lists over 200kB must be passed by URL”。请问这个问题应该如何解决?
我搜到“brranching”这个R程序包,应该可以处理大批量物种数据,但是“Phylomatic”这个命令只能处理小于5000的物种;而“Phylomatic_local”这个命令,在计算时需要安装命令行工具“awk”和“gawk”,但我不知道如何安装“awk”和“gawk”。不知道您对这个程序包是否有所了解?
回复 :
我之前建过比较大的进化树约16000个分类单元,是用Davies的科系统关系,用Phylomatic的本地版本创建的 (Phylocom软件所自带的Phylomatic.exe).
Phylomatic在线版本的限制比较多,对于分类单元的限制,目前我没有很好的解决办法。 Phylomatic的源代码托管在github上, https://github.com/camwebb/Phylomatic-ws 其中 \Phylomatic-ws-master\lib 下面的文件, 就是gwak语言的脚本, 用来处理进化树,拟合时间等。 不过我没有系统学习过网页编程, 所以看gwak的源代码还比较困难。
如果在本地使用网页版本的Phylomatic, 我觉得应该先安装Server, 一般来说, 是安装Apache Server, 同时要确保系统中有gawk的可执行文件,以便网页在执行时可以调用。在本地配置好之后, 再通过localhost访问。 安装Apache,如果是Windows, 则用Xamp最方便, 不过安装Gawk就没有那么直接了。 如果非要使用本地网页版本, 建议试试Ubuntu,Debian等Linux版本。这些系统安装Xamp以及gawk相对简单很多。
分类单元数非常多的进化树怎么建立? 提问 :
最近用1万种植物名,在Phylomatic网站建立系统发育树,两种方法都试过了,都不能正常运行:
taxa 中录入1万种的科属种时,结果显示: Trees/taxa lists over 200kB must be passed by URI
当我把科属种上传到网上URI, 输入URI之后,显示: More than 5000 taxa (too big). Sorry, please use an offline version of Phylomatic (see documentation).
回复 :
Phylomatic对输入的物种数是有限制的。可以考虑使用 浙江大学金毅编写的S.PhyloMaker。请参考
https://github.com/jinyizju/S.PhyloMaker https://doi.org/10.1093/jpe/rtv047
Phylomatic的进化树能获得bootstrap支持率吗? 提问 :
我用Phylomatic 建了一个系统发育树,想检测植物性状的系统发育信号,以及计算PIC和pGLS,来看系统发育对性状种间变异的影响。在投稿的时候,审稿人提出了一些问题,我不太懂,可否帮忙解答一下?
1)审稿人说我应该“report the bootstrap value”, 这个bootstrap值是自展值?这个数据我应该怎么获取呢?
2)审稿人希望“report which nodes are fixed”, 我问了系统发育方向的同学,说意思是化石标定点,我查了zanne et.al. 2014年的文章,查了里面的化石标定点,跟我选取的物种相关的标定点只有豆科那一支上有一个,这样可以吗?
回复 :
下面是我对问题的回复,供参考:
从Phylomatic重新获得的进化树是没有bootstrap value的。 只有通过DNA序列获得的进化树才能通过bootstrap 获得 supporting value。 审稿人非常清楚Phylomatic是如何获得进化树的。 因为Zanne在做进化树的时候, 应该是引用了很多化石点做进化树的时间校正,如果需要,可以引用Zanne的论文。
Phylomatic进化树和分子系统树的主要区别是什么?Phylomatic进化树能用于进一步分析吗? 提问 :
在一个学系统进化方向的同学的帮助下,我了解到,在Zanne的进化树上,有39个化石标定点,其中有一个是Fabales,我的进化树其中有一支是豆科植物,不知道这样算不算进化树上的节点与这些固定的化石有关系?
另外我的进化树中,有9个物种在Zanne的进化树上是可以找到的,另外的一些物种的进化时间可能是根据它说在你的属估算的。这样是否回答了“我的进化树上哪些节点在Zanne的进化树上是固定的”这个问题?
回复 :
Phylomatic建立进化树的方法, 主要是在科一级的水平上, 在backbone进化树上, 进行添加。 然后假设在科属种三个分类水平上, 分别延续了三分之一的时间 (Webb and Donoghue, 2005)。在属一级有信息是, 应该会按照属的时间添加物种(不知道是否是随机添加)。Phylomatic 主要用于群落和地区水平的物种系统发育多样性分析, 这主要是因为, 在Phylomatic发表的时候, 大部分物种的DNA条形码信息仍然是没有的,但是要近似了解物种之间的系统发育距离, 可以用Phylomatic提到的算法, 得出一棵进化树。 Phylomatic进化树与DNA条形码得到的进化树进行比较,在DNA条形码开始流行时, 已经有若干研究了, 主要是 John Kress团队(John Kress 的网站)以及裴男才博士等所做的工作。Phylomatic方法获得的进化树, 在研究系统发育距离较远的物种时,准确度是很高的。 但是如果有大量物种出现在同一个科属, 就会出现众多的多分枝结构, 影响对结果的计算。尤其是在PIC或者计算系统发育信号时。
参见Phylocom的说明书第26页 “To handle polytomies, AOT uses the method introduced by Pagel (1992) to obtain a single degree of freedom contrast at each polytomy, treating the polytomies as ‘soft’.”
参考:
这种情况下,我觉得是已经回答了审稿人的问题。 审稿人想让你列明哪些时间是在Zanne的进化树中是固定的, 哪些是你后来添加的。
分子系统树 多基因Supermatrix应该怎么构建? 提问 :
用多基因建树的supermatrix要注意什么?怎么建立supermatrix?
回复 :
利用多个基因建立系统发育树,有一种称为 supermatrix的方法。这种方法是将每个基因先比对好,再将比对好的序列,相同种的连接起来,其中空缺的位点用?替换,以此来构建一个超级矩阵。
由于不同基因,进化速率可能是不同的,因此需要在在软件中调用分隔模型,MrBayes、RAxML等,都是支持分隔模型的,设置分隔模型在这些软件的说明书中,都有详细的说明可以参考。
建立超级矩阵,可以使用R的phylotools程序包的supermat函数,不必管其基因名称,分别将片段命名即可。
关于Supermatrix的方法,也可以参考:Kress, W. J., D. L. Erickson, F. A. Jones, N. G. Swenson, R. Perez, O. Sanjur, and E. Bermingham. 2009. Plant DNA barcodes and a community phylogeny of a tropical forest dynamics plot in Panama. Proceedings of the National Academy of Sciences 106:18621-18626.
基于DNA序列的进化树应该怎么做? 提问 :
我有一个地区木本植物的名录,要建一个属水平的系统树,该怎么做?我是从genebank里查找序列,是只选择某一个种代表这个属还是要选择多个种?若选择多个种,怎样合并到一个属?选择哪些基因片段比较合适?若方便的话,还请传几篇这方面的文献。万分感谢!
回复 :
目前我了解到做进化树有两种方法:
一 从名录开始建立进化树,这主要是要用Phylomatic 和 Phylocom软件。
二 从DNA序列建树。也包含多种方法,一般用ClustalX或者MUSCLE等比对,用RAxML, BEAST, MrBayes等建立进化树,再用r8s或者别的方法进行分子钟校正。
用到的基因片断,目前有 rbcla, matk, its, trnh-psba等。可以参考 John Kress等人的几篇关于DNA-Barcoding的文献。据我所知:
rbcla 适合建立科和属以上的
matk 适合建立属和种水平的
its适合区分居群
trnh-psba适合区分居群
建立分子系统树的序列太多了怎么办? 提问 :
我尝试了建ML进化树,但是发现如此计算量计算机实在受不了,因为我有9000多条序列。有无比较快速的软件可用来建ML树的?我起初用FastTree是因为这个软件比较快速,而且所建的树是近似ML树。
回复 :
如果序列太多,那可能只能用NJ的方法做进化树了。还有一种方法:在 www.phylo.org 可以注册一个帐号,用那里的超级计算机,用RAxML等软件来建树。
有rbcLa、matK、ITS等DNA条形码片段了,应该怎样建树?怎样进行群落系统发育分析? 提问 :
有rbcLa,matK, ITS等DNA条形码片段了,应该怎样建树?怎样进行群落系统发育分析?
回复 :
有DNA条形码序列之后, 请按照下面的方法建立进化树:
将rbcLa,matK, ITS等片段分别用 比对 (MAFFT或者MUSCLE)
用phylotools程序包/evobir 程序包或者biopython或者Genious 合并成 supermatrix,也就是合并了多个比对好片段的phylip文件
用RAxML,或者IQTREE www.iqtree.org/ 建立最大似然进化树
用r8s或者PATHd8进行分子钟校正,枝长单位一般为百万年,参考 https://github.com/helixcn/EcoEvoLinks。
用Phylocom或者picante软件计算群落系统发育结构
建立进化树和相关的分析, 可参考以下网站: http://ib.berkeley.edu/courses/ib200/ http://treethinkers.org/ https://www.plob.org/article/2193.html https://www.geneious.com/features/phylogenetic-tree-building/ http://kembellab.ca/r-workshop/biodivR/SK_Biodiversity_R.html
希望里面的培训材料对你有帮助。
PD 系统发育多样性 怎样用Phylocom计算系统发育多样性? 提问 :
我想用Phylocom计算PD指数,现在的问题是,输入命令“Phylocom pd result.xls”后,系统就报错“Phylocom.exe遇到问题需要关闭……”,试了多次均如此。我是用的FastTree软件建的树,用记事本打开后发现也是Newick格式的树。据我有限的知识,也看不出格式有哪里不对。我已经将sample和phylo文件放在附件中,麻烦帮忙看看问题在哪里。
回复 :
您的sample文件是xls文件? sample文件必须是纯文本文件,并按照Phylocom的要求准备。
Phylocom计算所需要的进化树必须是Ultrametric进化树。我在Figtree中查看了一下您传给我的进化树,发现并不是UltraMetric的,所以必须要经过转换。但是您传给我的可能是Bootstrap之后,经过合并的,这样对计算结果影响很不好。我一般是用RAxML建立ML的进化树,而且只建立一棵,不进行bootstrap运算以及合并等。
我用如下命令,将您传给我的进化树转换成了Ultrametric Tree,这在r8s等软件中可以实现。
1 library(ape)setwd("C:/")tre <- read.tree("phylo")tre2 <- chronoMPL(multi2div(tre))write.tree(tre2, "new.tre")
可以试试以上几句R的命令, 不知是否能解决您的问题
怎样去掉PD里面物种丰富度的影响? 提问 :
想问一下怎么把 phylogenetic diversity(Pd) 中消除Species richness 的影响?有什么指标代替标准化的pd 吗?
回复 :
SES PD 我们应该是用picante程序包的 ses.pd直接计算出来的。
请参考
1 library(picante)?ses.pd> data(Phylocom)> ses.pd(Phylocom$sample, Phylocom$phylo, null.model="taxa.labels", runs=99) ntaxa pd.obs pd.rand.mean pd.rand.sd pd.obs.rank pd.obs.z pd.obs.p runsclump1 8 16 26.31313 1.607676 1.0 -6.4149319 0.010 99clump2a 8 17 26.17172 1.708564 1.0 -5.3680843 0.010 99clump2b 8 18 26.27273 1.608765 1.0 -5.1422831 0.010 99clump4 8 22 26.58586 1.498367 1.0 -3.0605707 0.010 99even 8 30 26.50505 1.567357 100.0 2.2298368 1.000 99random 8 27 26.64646 1.416471 58.5 0.2495888 0.585 99>
为什么PD跟纬度负相关,而SES.PD和纬度正相关? 提问 :
谢谢您的回复!我还有个问题,前面算过 ses.pd ,但本来PD跟纬度负相关,但算出来的ses.pd发现跟纬度正相关,这个应该怎么理解?谢谢!
回复 :
pd和物种数是有极显著的正相关关系的, 而ses.pd 只是比较pd和零模型所得pd的差别, 如果一个地区本身pd值很高, 则ses.pd的值也应该是很高的。 另外,纬度越低物种丰富度就越高, pd就越高, 同时pd.ses也应该是高的。 因此我也推测纬度越低, ses.pd应该越高。所以一定是负相关的关系。
如果真的出现了您所说的正相关关系, 还应该从取样的偏差, 甚至是ses.pd函数本身是否存在bug等方面考虑。
没有枝长的进化树,能计算PD吗? 提问 :
我用Phylomatic生成了一棵树,后面用Phylocom可以算出pd,但是放在R里面算时,提示说Tree has no branch lengths, cannot compute pd。Phylomatic生成的树不是应该带枝长的吗,是怎么回事呢?
下面是我用的命令代码:
1 install.packages(c("picante"))setwd("C:/tree")library(ape)library(picante)phylo <- read.tree("phylo.txt")sample <- read.table("sample.txt")pd <- ses.pd(sample, phylo, null.model="taxa.labels",run=999,iterations=1000)write.csv(pd, file="SES.PD.csv")
回复 :
计算PD用的进化树, 是需要有枝长的。否则在计算一个种到另一个种的系统发育距离时,只能计算节点的数量了, 但是节点数直接加减其实并没有多大的意义。
您的脚本,我发现两个问题:
进化树文件在最后没有英文分号 ;, 所以在read.phylo时会出现问题。这是跟newick文件的定义有关。
Phylocom sample文件不能在picante中直接使用, 而是要先转换为matrix, 用 sample2matrix函数可以进行相应转换。
修改后的R脚本如下。
1 #### install.packages(c("picante"))setwd("C:\\Users\\jlzhang\\Desktop\\attachments")library(picante)phylo <- read.tree("phylo.txt")phylosamp<- read.table("sample.txt")mat <- sample2matrix(samp)pd<- ses.pd(mat, phylo, null.model = "taxa.labels",run = 999,iterations = 1000)write.csv(pd, file="SES.PD.csv")
怎样计算PD系统发育多样性计算? 提问 :
我基于ITS序列构建了系统发育树(Mega软件)。接下来我想计算每个群落内物种的谱系多样性。不知道怎么计算?能用R吗?您方便能指点一下吗?
回复 :
目前很少有单基因建立进化树了。最好考虑用多个基因建立进化树。
要计算系统发育多样性,应该先获得ultrametric tree, ultrametric进化树一般是通过分子钟校正获得的,枝长表示分化时间, 一般是r8s或者BEAST, 也可以用PATHd8等软件来做。获得ultrametric 进化树后, 再用Phylocom或者picante计算系统发育多样性就可以。具体请参考Phylocom或者picante的说明书。
零模型 零模型的置信区间 提问 :
通过Phylocom中Independent swap作为零模型,我计算出了NRI和NTI,请问有没有一个置信区间,超过这个区间就是谱系聚集,低于这个区间就是谱系发散,在这个区间就是谱系随机呢?这个有文献吗?谢谢
回复 :
Nate Swenson 博士曾经提到,NRI以及NTI都是standard effective size, 在95% 的置信区间, 如果数值超过1.96或低于-1.96, 都属于显著。 但是这有一个前提, 就是零模型生成的MPD或者MNTD呈近似正态分布。Swenson博士的论文中应该提到了这一点。
怎样检测性状的空间分布是聚集还是分散? 提问 :
我想知道某一功能性状(例如LMA)在群落中的分布,比如是聚集的或是分散的?该怎样构建一个零模型(null model)呢?怎样通过R或者其他软件怎样实现?
回复 :
性状在群落中的分布, 也是可以按照群落系统发育相似的手段进行分析的。
在R软件中, 先将性状进行标准化后, 计算不同物种同一性状的欧几里得距离,用UPGMA或者Ward方法等,进行聚类分析, 构建一个不同物种, 同一性状的聚类树(hclust类型的数据), 然后用将其转换为phylo类型。之后, 就可以参考picante程序包, 以及利用相应的零模型, 研究性状是聚集或者发散的。
零模型,是群落中出现的物种进行随机化,零模型与我们要检验的假说有关。群落系统发育中常用的有四种, 具体参考Phylocom的说明书。
样方数量会影响零模型的结果吗? 提问 :
如果要做1ha森林样地不同 spatial scale 群落谱系结构,1010、2020、5050,按照样地的坐标选择,则小样方分别有100个、25个、4个,最后进行谱系关系指数的计算,那么50*50的小样方只有4个,这样的结果会不会没有可信度?
回复 :
这取决于您使用了哪个零模型。Phylocom的 tip shuffling 零模型只是将物种的系统发育顺序随机打乱, 结果一般都不会受到样方数很大的影响。但是其他零模型就可能会受到影响。 请参考Phylocom说明书 第6.1.3小节
能否用零模型检验样方物种组成在多大程度符合中性理论的预测? 提问 :
ses值只能检测群落的聚集和发散两种情况,那对于中性理论中存在的随机情况,如何进行检测?
回复 :
Phylocom的零模型, 就是在用不同的方式模拟随机情况,作为standard effective size, SES >1.96 或者 SES < -1.96 即可认为在 95%的水平上显著, 也就是存在显著的聚集或者发散。 如果SES介于两者之间, 则很大程度上, 受到随机过程的影响。但是现在并未找到很好的办法量化随机过程在其中能解释多少方差。
零模型中的零分布是什么? 提问 :
做植物群落系统发育分析应该看过Bryant 2008年发在PNAS上面的那篇文章(”Microbes on mountainsides: Contrasting elevational patterns of bacterial and plant diversity”)。这里面我有个问题我一直没有弄明白的。
在Materials and Methods部分,Compositional and phylogenetic similarity章节中,作者提到 :
“We tested whether the slope in the decay of phylogenetic similarity was greater or less than what was expected given the taxonomic decay in similarity by comparing the observed slope with a distribution of distance–decay slopes obtained by randomizing the location of taxa at the tips of the community phylogenetic tree 1,000 times. This is equivalent to randomly sampling the taxa while constraining the number of taxa in each community, the number of taxa shared by any two communities, and taxa occurrence across all communities. The observed phylogenetic slope was assumed significantly different from the null if it was greater than or less than 975 of the slopes of the randomizations (two-tailed test, P < 0.05). ”
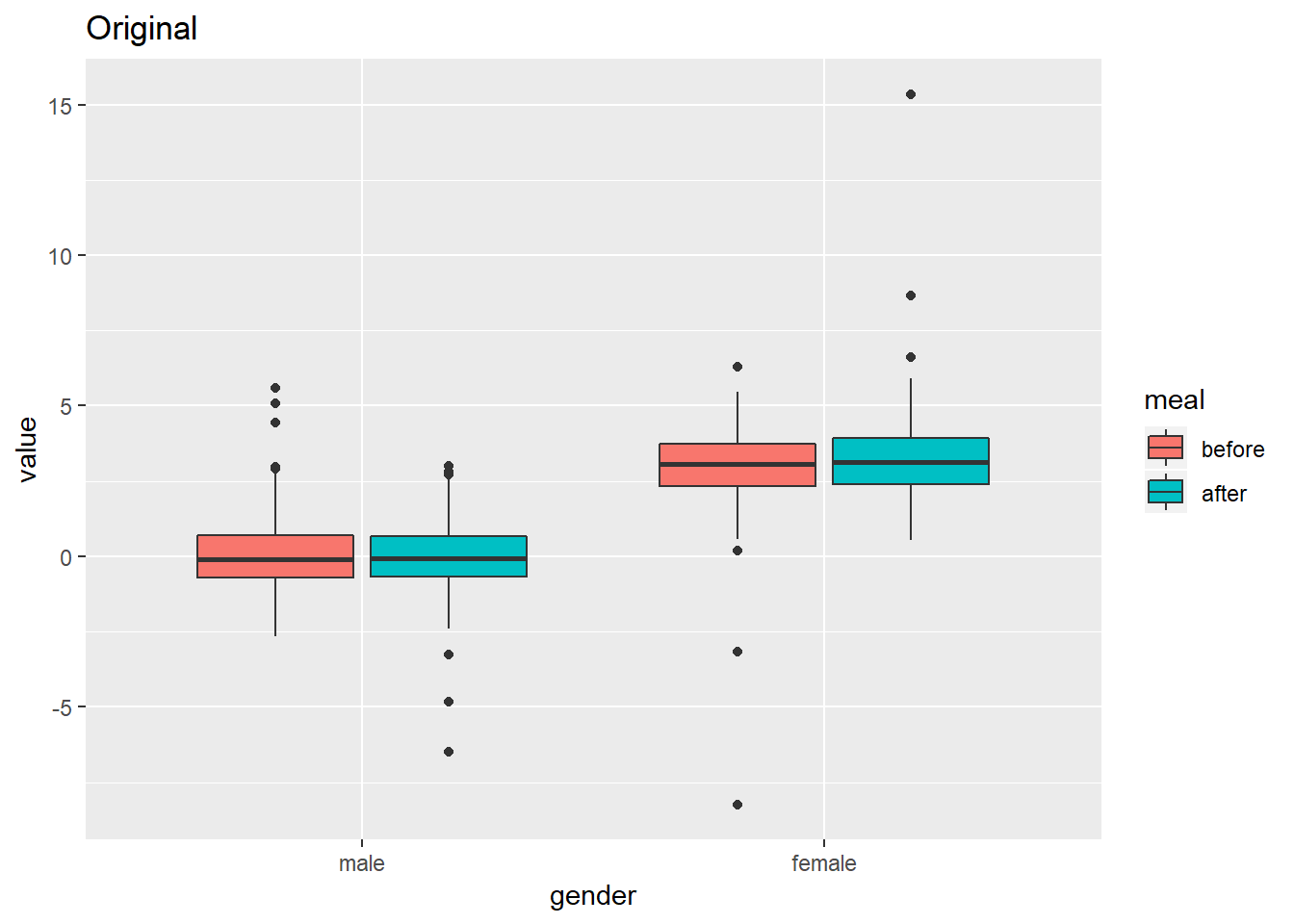
我很想知道,作者是如何得到在null model下的随机化结果?仅仅是一次随机化吗?如果不是单纯一次随机化,如何计算他们的Null phylogenetic Similarity?就是下图中的结果。
回复 :
作者已经提到是随机化1000次, 每次随机化, 都是对进化树(对tip label进行随机重排)进行随机化。然后针对每一棵进化树, 都进行类似的计算。 这1000次计算得到的结果, 就是null distribution. 按照从低到高的顺序(或者从高到低)进行选第975个值(或第25个的值)作为临界点(P<0.05, two tails test),与真实值进行比较。以近似得获得显著性。
如何自己在R中创建群落随机化零模型? 提问 :
我看很多文献中,在不考虑系统发育的情况下,可直接计算一个样地alpha或者两个样地beta的多样性;这样的话直接同自己用R构建的null model比较,计算效应值(均值差/sd);想问下我如果想用R构建样地的null model要怎么做?您能否给我一个模板文档?还有关于两个样地差异的null model同一个样地内的物种随机,R程序是否存在很大差别?
回复 :
请参考如下命令
1 library(picante)dat <- read.table("sample", header = FALSE)datmat <- sample2matrix(dat)###### By Site rndr_datmat <- datmat[sample(1:nrow(datmat)),]###### By Specierndc_datmat <- datmat[,sample(1:col(datmat))]
什么时候要考虑零模型? 提问 :
在看到很多文章中提到NRI的提取是用Phylocom软件或者R语言,而大多数文章中没有提到零模型的问题。您认为什么时候该考虑零模型假设的问题,什么时候不必考虑呢?
在分析系统发育结构特征与环境变量的相关分析中,如果在某个分类阶元上物种种类较少,这样的分析是否可行?或者说这样的分析会有什么问题存在?
回复 :
在看到很多文章中提到NRI的提取是用Phylocom软件或者R语言,而大多数文章中没有提到零模型的问题。您认为什么时候该考虑零模型假设的问题,什么时候不必考虑呢?
所谓零模型, 就是群落数据或者进化树数据进行随机化的方式。 运行零模型主要是为了解, 在随机状况下, 物种系统发育关系的统计分布,从而推断真实的群落内物种的系统发育关系是否随机, 以便了解environmental filtering或者stochastic process的相对重要性。所以在探讨这些生态学过程的相对重要性, 计算NRI或者NTI的时候, 必须要考虑零模型。
NRI的计算,必须要涉及到零模型, 而目前Phylocom中零模型有四个:
Phylogeny shuffle:
Species in each sample become random draws from sample pool
Species in each sample become random draws from phylogeny pool
Independent swap
picante 程序包中, 零模型有7个
taxa.labels: Shuffle distance matrix labels (across all taxa included in distance matrix)
richness: Randomize community data matrix abundances within samples (maintains sample species richness)
frequency: Randomize community data matrix abundances within species (maintains species occurence frequency)
sample.pool: Randomize community data matrix by drawing species from pool of species occurring in at least one community (sample pool) with equal probability
phylogeny.pool: Randomize community data matrix by drawing species from pool of species occurring in the distance matrix (phylogeny pool) with equal probability
independentswap: Randomize community data matrix with the independent swap algorithm (Gotelli 2000) maintaining species occurrence frequency and sample species richness
trialswap: Randomize community data matrix with the trial-swap algorithm (Miklos & Podani 2004) maintaining species occurrence frequency and sample species richness
2.在分析系统发育结构特征与环境变量的相关分析中,如果在某个分类阶元上物种种类较少(例如,以白腹鼠属为研究对象,而只有6个种),这样的分析是否可行?或者说这样的分析会有什么问题存在?
这里的系统发育结构指的是什么? 如果你的species pool种类非常少, 则randomization所能提供的排列组合数是非常有限的。会丧失statistical power。从小样本推断总体, 如果是基于随机化方法, 都会面临类似的问题。
系统发育信号 检验系统发育信号时, 进化树必须是超度量树吗? 提问 :
检验系统发育信号要注意什么?是否要求必须是一棵超度量树(带枝长的,有分化时间的树)?
回复 :
在检验系统发育信号, 进行群落系统发育时, 进化树必须是ultrametric的, 即右侧为等距的(超度量树), 其枝长表示时间。不能直接用RAxML,PAUP*,PHYLIP,PHYML,GARLI,MrBayes等软件直接生成的进化树,因为这些软件生成的是ML树或者MP树,枝长并不表示分化时间。BEAST软件生成的进化树, 枝长表示时间, 可以使用。 枝长不是时间的进化树, 必须通过分子钟校正或者一些转换, 转变为ultrametric tree。 转换的方法, 可以用r8s, PATHd8,multidividetime, ape程序包的chronopl等。
功能性状数据检验系统发育信号时, 需要做log转换吗? 提问 :
我们采用形态特征数据检验系统发育信号时,是否需要将原始数据(原始数据单位为cm)进行 log transformation 的转换?
回复 :
建议进行转换,Blomberg, S. P., T. Garland, Jr., and A. R. Ives. 2003. 的论文中,对数据进行了转换。但是是否需要进行转换, 我并未见过什么标准。
计算Blomberg’s K时, 怎样看待其对应的p值? 提问 :
K值和p值二者,究竟以哪个结果为标准检测系统发育信号?当p值显著,k值小于1的情况,如何定论?
回复 :
系统发育信号, 用K来表示。 由于k 只是一个比值, 表示的是相对大小。 K大于1,表明物种的性状比按照布朗运动出现的组合要近。反之要远。由于你的k值都小于1, 因此应该认为, 系统发育位置相近的种, 它们的性状比布朗运动模型预测的要远。 P是否显著, 只是对K的显著性进行检验, 一般采用tip shuffling , 将标签随机排列, 这样物种的系统发育关系在进化树上随机排列,可以检验K相对于这个零分布是偶然的, 还是必然的(P < 0.05).
存在系统发育信号,就是性状“系统发育保守”吗? 提问 :
存在系统发育信号,就是性状“系统发育保守”吗?不存在系统发育信号,就是性状“系统发育趋同”吗?
回复 :
如果K大于1,就说存在系统发育信号 , 表明系统发育位置相近的物种, 具有相似的性状。 如果K小于1 ,可以说存在系统发育反信号 (Anti Phylogenetic Signal ) , 此时, 系统发育关系相近的物种反而具有更加不同的性状。 若K的检验都不显著, 则可以认为, 不存在系统发育信号, 但这并不代表“系统发育趋同”。 你提到的“系统发育趋同”, 可能这里说的反信号。 如果不存在系统发育信号, 也就是性状的进化符合布朗运动模型所做出的预测(零分布)。
Phylocom Phylo文件中Segmentation fault: 11错误 提问 :
我的phylo和sample文件在附件中,使用命令为Phylocom comstruct -m 0 >output.txt,程序总是提示Segmentation fault: 11错误,经反复找错与测试,我发现是phylo文件有问题,可我不能确定phylo文件哪里出了错。还望能够帮忙
回复 :
数据有两个问题:
sample文件中, 只有一个样地的数据。两条数据,这个数据量太少, 无法运行 comstruct
phylo文件不含有枝长。 如果不包括枝长,Phylocom会假设所有枝长为1,进行相关计算。但是由于此时进化树会变成 non-ultrametric ,枝长不能加减,因此结果无意义。
Phylocom能否分析群落遗传多样性? 提问 :
用Phylocom软件是否可以进行群落内的遗传多样性相关分析?
回复 :
Phylocom不是用来分析遗传多样性的,主要是用来分析系统发育多样性和系统发育结构的。
检查sample和phylo文件时,怎样删除文本文档中的非ASCII编码字符? 提问 :
纯文本文档中有ascii编码的符号只能一个个检查?是否可以通过某些软件直接转换保存?
回复 :
可以用notepad++编辑器打开文本后,在允许正则表达式的情况下替换全部非ASCII字符。[^\x00-\x7F]+
样方中没有出现的物种,不要列在sample文件中 提问 :
我用45个样方数据,准备了phylo文件和sample文件,但我不知是什么地方出了错误,所计算的PD值和系统发育距离全是一样的值。我将Phylo文件和sample文件以及计算结果都上传到附件了。
回复 :
您的sample文件有问题,个体数为0的物种应该全部删除。所有的其他指数都是以PD为基础的, PD计算得不准确, 其他指数也就不可能准确。 更多指数计算,请参考 picante程序包 http://blog.sciencenet.cn/home.php?mod=space&uid=255662&do=blog&id=1116212
sample文件时可不可以用科或者属名来当样方名、直接用1来当所有的物种数呢? 提问 :
我正在尝试用Phylocom计算我的系谱树中的NRI和NTI,应该需要用到comtruct,但是不太清楚怎样构建sample文件……我手头只有一个dated tree,没有样方和物种数相关的资料,这样的情况下请问制作sample文件时是不是用科或者属名来当样方名、直接用1来当所有的物种数呢?还有,请问您博客中提到的
1 phylodist <- as.dist(cophenetic.phylo(tr))
是否就是计算mean pairwise distance呢?
回复 :
sample文件, 应该按照Phylocom的要求构建。sample文件是物种在某个样方出现情况的纪录。
该文件内容必须为纯文本
该文件分为三列, 各列之间, 用制表符间隔开(可以通过Excel等软件, 另存为txt)。
该文件没有表头,
第一列表示样方的名称, 样方的名称不能有空格
第二列表示个体数, 必须为整数
第三列, 表示物种名。 物种名必须出现在进化树中。 如果没有的话, Phylocom不能运行。
以上就是sample文件的要求。
怎样制作sample文件?群落系统发育能研究物种入侵吗? 提问 :
假如我想研究北京整个地区的群落组成,我就直接把北京看成一个整体行吗?还是必须要把北京划分成许多个亚地区?
这个sample文件一般是怎么得到的呢?是自己实地调查的结果?还是根据标本的记录?
sample文件中有一列信息是物种的多度,指的是该物种在分布区的数量吗?
您之前说MPD和MNND可以对群落的物种组成机制做推断,我最近看的文献是把这种方法运用于外来入侵植物的研究,不知您是否有关注?您觉得有什么区别吗?
因为我确实不太了解生态方面的问题,而且还没有实际运算过数据,所以问题比较多,再次感谢您的解答。
回复 :
这取决于研究的问题。 我并不太清楚您所研究的问题。
在群落尺度, sample文件,多度是物种的个体数。 Phylocom说明书已经写了, 怎样生成sample文件。在地区尺度, sample文件一般是物种名录。 很显然, 名录既可以通过调查获得, 也可以通过标本记录获得。
多度, 这里只是个体数, 即某一个样方中, 有多少个个体等,与分布区数量无关。
很久之前Sharon Strauss分析过入侵物种会改变群落的系统发育结构。 你可以检索一下 community phylogenetic structure, 文献很多。 我并不是做入侵物种的研究。 我觉得要做什么分析, 还是要跟从要回答的问题出发。
sample文件:Mixed delimiters in file:sample 提问 :
在运行Phylocom计算betaMNTD的时候,输入Phylocom comdistnt>comdistnt output.txt命令,软件提示错误,显示:Mixed delimiters in file:sample。不知道哪弄错了,还望指点!原始数据在附件里面,谢谢您了!
回复 :
数据有两个问题:
phylo文件为newick格式的进化树没有错, 但是不是ultramtric的, 可以尝试用R ape程序包的 chronos 函数转换为ultrametric格式的进化树,才能进行计算,否则得出的phylobeta或者PD等都是没有意义的。
sample文件可以通过excel格式另存为 txt格式, 制表符间隔即可
sample文件是必需的吗? 提问 :
我想用这个软件计算PNND和MPD时,必须需要sample文件吗?如果只有一个phylo文件的系统发育树可以计算吗?我有点不明白,为什么计算这两个谱系距离需要物种分布信息(sample)?
回复 :
我不不清楚PNND是什么,MPD应该是 mean phylogenetic distance。
Phylocom软件在计算MPD时, 需要有phylo文件和sample文件。
sample文件的要求请参考 Phylocom说明书的第9页, 3.2节 (https://github.com/Phylocom/Phylocom/releases/download/4.2/Phylocom_manual.pdf )。 MPD可以在计算NRI指数时获得。如果只有进化树,那么可以准备包含所有种的sample文件, 每个种的abundance都是1即可。
提问 :
一般计算遗传距离不是只需要矩阵或者发育树就可以了吗?为什么使用Phylocom计算MPD(Means Phylogenetic Distance MPD)分类单元间平均谱系距离和 PNND/MNND(phylogenetic nearest neighbour distance) 最近相邻谱系距离,需要物种分布信息呢?
回复 :
MPD是某地点的系统发育平均距离。
举例: 假设现有A和B两个地点, A地点所包含的系统发育多样性在平均以后, 是否比B点要高? 这时候就需要A 和B两个地点的物种名录。分别计算A地点内所有种两两之间(两两之间成对)的系统发育距离,再除以两两之间的对数(number of pairs)。B地点与之相同。
PNND/MNND(phylogenetic nearest neighbour distance) 在Phylocom中称为MNTD。对于地点A的种,假设有sp1, sp3, sp8…spn个种,只计算在系统发育距离上,每个物种与其系统发育距离最近物种的系统发育距离,再平均以后就得到了A地点的MNTD。B地点计算方法与之相同。
MPD和MNTD主要是为了探讨群落内物种的平均系统发育距离与零模型相比是高还是低。从而对群落组成机制做一些推断。
为什么会出现Taxon XXXX in sample not a terminal taxon in Phylomatic. exiting 提问 :
运行 Phylocom comdist > comdist.output.txt 命令后,结果显示"Taxon Acer in sample not a terminal taxon in Phylomatic.exiting."是不是我改成自己的sample文件时候用错了呢?
回复 :
sample文件中的物种名与phylo文件中的物种名必须一一对应, 包括大小写。
怎样准备sample文件?MPD.rankLow和MPD.rankHi是什么? 提问 :
我想使用软件中的comstruct和nodesig功能,一个问题是在准备sample和traits文件时,sample文件中的第二列多度的含义以及traits文件中的性状数据信息我不知道该怎么定义或是填写。
另一个问题是,我尝试使用了comstruct功能,对得到的结果文件中的MPD.rankLow和MPD.rankHi这两列想不通,就像我下边的截图所示,第一列随机的MPD比实际的MPD要低,为什么MPD.rankLow会比较低?我的理解是随机的MPD既然比实际的低,那么出现MPD.rankLow的次数应该要多,我不知道是不是我的理解有误,实在没想通这个地方,以及算单尾p值的时候是不是直接用MPD.rankLow除以1000次?
回复 :
请参考Phylocom说明书第9页:
3.2 Sample preparation
3.3 Traits preparation
说明书 15页
The rank of observed MPD/MNTD values relative to the values in the null communities are reported as rankLow (number of null communities with MPD/MNTD values less than or equal to observed) and rankHi (number of null communities with MPD/MNTD values greater than or equal to observed).
rankLow是将真实的MPD与每次零模型随机化生成的MPD放在一起, 一般情况下, 假设设置999次零模型, 那么加上真实的MPD, 则共有1000次计算。 将数值按照从大到小排列, 数值高低的序号称为rank。rankLow定义为:零模型生成的值比真实值小的序数,在你的结果中是998个随机值比真实值要小。
用Phylocom能建立群落聚类树吗?为什么picante会下标出界? 提问 :
我知道在“picante”里面计算comdist.result后,可进一步聚类,但用Phylocom得到的群落两两系统发育距离,怎么能用于构建群落的聚类树?我用“picante”能够计算出PD,但是在计算其它值时,出现“下标出界”,后面就运行不了了。我就想着用Phylocom的结果来聚类!
回复 :
如果已经有样方之间的系统发育距离矩阵,或物种组成相异性矩阵, 都是可以进行聚类分析的, 以反映哪些样方之间的系统发育关系相近或者物种组成相似。 可参考 http://ecology.msu.montana.edu/labdsv/R/labs/lab13/lab13.html
picante中“下标出界” 的问题, 主要是有些物种在群落数据中出现了, 但在进化树中没有出现。 注意R中, 大小写要完全对应的, 学名一定要完全匹配。 在进化树中没有出现的物种, 需要在数据处理时 从community matrix中去掉。
Taxon in sample not a terminal taxon in phylogeny? 提问 :
我现在想做群落两两之间的系统发育距离,但是用Phylocom comdist进行运算时,数次尝试的结果都是只有一句话:Taxon in sample not a terminal taxon in phylogeny. Exiting.我检查了我phylo文件和sample文件中的物种名,二者也是相对应的,并没有不一样。所以想请您帮我看一下问题出在哪里。
回复 :
sample文件也必须是纯文本格式, 一般是用excel另存为 txt 文件, 然后去掉扩展名。 您的sample是xlsx格式的, 我已经转换为txt格式了, 并且计算成功。结果参见附件。
另外, 值得注意的是, 进化树需要有枝长, 而枝长一般是通过Wikstroem的分化时间拟合的。 而您的进化树目前没有枝长。 所以还要用BLADJ或者别的方法, 为进化树添加上枝长。
提问 :
“ 输入 Phylocom bladj > out.tre 生成out.tre文件,就是含有枝长的进化树。”在这里总是提示说Phylocom.exe程序出现问题,无法进行后面的操作。你提供的软件版本是4.1,但是现在是4.2版本,是否为这个原因?期盼你的回复!
回复 :
可以先看一下, phylo文件是否有非ASCII码的字符。
提问 :
现在有一些关于进化树的问题想请教您,我用Phylomatic建的进化树,用Phylocom 拟合出的枝长,发现一些不同属不同种的枝长都是一样的:
拉丁名
枝长
树参
Dendropanax_dentiger
11.206897
变叶榕
Ficus_variolosa
11.206897
九节
Psychotria_asiatica
11.206897
鹅掌柴
Schefflera_heptaphylla
11.206897
我想问您出现这种结果的原因是什么?
回复 :
这是和Phylocom BLADJ的算法有关的。 如果有科的分化时间, 则BLADJ会将科的时间三等分。 如果没有科的分化时间, BLADJ会继续将更高等级的节点继续等分。如果你用的是Wikstrom的分化时间, 则Araliaceae的分化时间是26Myr, 而Moraceae为23.0Myr, 请问你是否用的 Wikstrom的分化时间? 还是用的别的分化时间?
为什么CMD中会提示“Phylocom不是内部或外部命令” 提问 :
在开始> cmd > cd C:\Phylocom>输入Phylocom comstruct > comstruct.output这一步中按开始,运行输入cmd出来的黑色框中,我输入了C:\Phylocom,它就出现C:\Phylocom不是内部或外部命令,也不是可运行的程序或批处理文件,是我的步骤错了还是什么?
回复 :
由于Phylocom.exe是 console application,可以添加到system path,再通过CMD运行,也可以解压缩在某文件夹之后, 再在相同文件夹内创建一个.bat文件运行, .bat文件里面放上相应的命令,如Phylocom comstruct > comstruct.output 即可。
Phylocom能计算植物以外类群的系统发育多样性吗? 提问 :
想请教一下您,Phylocom能做鱼类谱系方面的研究吗?在谱系树的建立中自己就遇到了很大困难,根据里面phylo文件的格式自己编写了要研究海域的编码,但对bladj、ages、sample等文件还是遇到很多问题,这些文件代码都是关于植物的。希望您能够帮忙解答一些疑问,期待您的回复!
回复 :
Phylogenetic structure是从研究森林群落开始的,这方面的研究比较多些。 Phylocom和Phylomatic也主要是基于植物群落做分析的。 不过Phylocom可以分析有物种名录以及进化树的任何群落的系统发育结构。而对于鱼类来讲,相信进化树是有的, 需要获得的只是群落的物种组成,也就是某一个地点鱼类的群落系统发育结构, 附件中是一些文献, 供参考(略)。
系统发育beta多样性 关于Unifrac 提问 :
Unifrac指数最早用于微生物, 那么:
是不是这个指标用到植物方面有问题或者不太适合植物?
看植物beta多样性中系统发育的组成,是不是Unifrac这个指标比MNTD更好一些。在用系统发育性Beta多样性指标时候,Unifrac与MNTD侧重点有什么不同?
回复 :
Unifrac指数最先用于微生物组成的系统发育多样性中。该指标用于植物群落的系统发育beta多样性并无不妥, 但是Unifrac和Phylosor的形式是类似的, 表达的信息也相若。
Unifrac用的还是比较多的。 描述物种的beta多样性, 有多度(个体数)信息时候用Bray-Curtis Distance, 没有多度的时候用Jaccard Distance。 描述系统发育多样性,可以选择的多样性指数包括 MPD、MNTD、Phylosor、 Unifrac等,其中MPD以及MNTD都可以选择是否进行多度加权。 在我看来, MNTD损失的信息比较多, 因此用来描述phylo beta diversity可能并不合适。 MNTD主要是探讨近缘种共存的。 所以使用Phylosor以及Unifrac都是一样的。
在上述几个指标中, 只有MPD比较独立于species beta diversity,但是引入MPD以及MNTD的最初目的, 都是为了计算群落的系统发育结构。 MPD, MNTD, PhyloSor 以及Unifrac这几个指数, 是无法完全独立于species beta diversity的。
Fabien Leprieur 似乎有探讨过这个问题,参考 http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0042760 。
怎样计算betaNTI? 提问 :
我用Phylocom计算betaNTI时出现了空文件,之前已经算出alphaNTI了,这就说明phylo和sample文件是争取的,我想请问您,是否可以帮我算一下betaNTI?谢谢!
回复 :
使用以下命令就可以计算出betaNTI和betaNRI了。
1 Phylocom comdist -f=phylo -s=sample>res_beta_nri.txtPhylocom comdistnt -f=phylo -s=sample>res_beta_nti.txt
物种数太多时,怎样计算系统发育beta多样性? 提问 :
物种数太多,如何计算phylogenetic beta diversity?
回复 :
sample数据的行数太多,可考虑用 PhyloMeasures 程序包 (我对这个程序包也不太了解),参见 Tsirogiannis et al. (2016)的论文。
Tsirogiannis C, Sandel B. Fast Computations for Measures of Phylogenetic Beta Diversity. PLoS One. 2016;11(4):e0151167. doi:10.1371/journal.pone.0151167
系统发育beta多样性的显著性是怎样检验的? 提问 :
我在使用Phylocom软件计算betaNTI时,发现数据结果的并不是对角矩阵,是不是要把矩阵的对角全部替换成0,在以此基础进行?
我用model 中的数据Phylocom comdistnt –s sample -f phylo -m 0 -r 999 -a -n>nti.txt同时我在很多文献中讨论中也没有具体说群落betaNTI值的是否存考虑权重 -a?
在计算效应值时,构建的零模型是否要考虑限制和非限制的区别?:
1 $ Phylocom comstruct -m 0 -r 9999 -a :
选择随机shuff( 重排系统发育关系和样地物种,这两个有什么偏好选择?)
$ Phylocom comstruct -m 3 -w 100 -r 999 : NTI comdist: 主要是对角值是否要替换成0?进入下一步计算?
附件是我用模型数据做的betaNTI结果。 其中是不是betaNTI的显著水平确认是不是只有绝对值小于2的情况才是中性过程?
回复 :
计算betaNTI时, 使用的是mntd,其实是寻找样方中,与某一物种最短的进化距离, 这个距离是不能用0代替的。所以不能将对角变为0。
我使用sample以及phylo文件, 按照以上命令, 运行得到了一样的结果。
很多情况下讨论betaNTI的值时,很多数据只有植物名录, 此时没有办法考虑物种多度加权的。 既然你的数据有多度, 则应该在计算的时候考虑,至少作为附录。
我觉得第3号模型 Independent Swap并没有特别适用的情况。 可能还需要阅读一些这方面的文献, 以考虑什么理论假设成立的情况下, 应该用Independent Swap模型。 不能换成0, 但是如果comdistnt的结果只作为系统发育beta多样的性的度量,则可以不必考虑群落自己与自己的系统发育多样性距离。
如果按照中心极限定理, NTI或者NRI的值, betaNTI以及betaNRI的值, 都应该接近标准正态分布, 此时, 在95%的置信区间,临界值分别为 -1.96以及+1.96, 小于-1.96, 以及大于+1.96, 都是偏离于95%的置信区间的, 也就是您说的显著。 如果落在这个区间之内, 则不能排除中性过程 (因为中性过程这里简化为随机, 如果真实值与随机生成的数据没有显著分别, 则不能排除真实值是随机生成的)。
怎样计算phylobeta diversity并绘图? 提问 :
如何在R中获得Null model下的MPD以及phylobeta diversity的参数值(例如phylosor)。就像您说的,在R中可以获得MPD每次随机化以后的值,如何获得呢?
最近在网上找到您很早以前分享的一个PPT文档《谱系多样性与分析方法》,能不能介绍一下如何在R中得到谱系beta多样性随空间距离、降水差异的关系图?这是否是两个矩阵之间的相关性分析?应该用哪个package和fuction来计算?
回复 :
我的理解是, 只能通过编程获得, Phylocom和picante中算法中都已经包括了,主要是在C语言中算的,不能得到每次随机化的值,因为得到了也是为了获得最终结果,而我们关心的其实只是结果。如果确实需要,可以更改picante中的相应C代码和R函数,以获得所需的值。
系统发育beta diversity是 将 系统发育距离矩阵和地理距离矩阵, 环境因子差异矩阵进行回归之后形成的。 可以参考 simba package:liste。
Rao二次熵 怎样计算Rao二次熵? 提问 :
NRI和Rao二次熵分别需要准备什么格式的数据源?
tre格式谱系树用R如何提取上一步所需的数据源?
使用R中的FD、picante程序包具体进行NRI和Rao二次熵两个参数的计算?
回复 :
①NRI和Rao二次熵分别需要准备什么格式的数据源?
NRI只能通过Phylocom软件来计算, 格式请参见Phylocom软件的说明书, 需要sample和phylo文件, phylo文件为newick格式的进化树, sample文件为Phylocom指定的格式。 Rao’s Quadratic Entropy 可以用Phylocom软件计算, 也可以通过picante计算。
②tre格式谱系树用R如何提取上一步所需的数据源?
上述软件所用的进化树, 就是newick格式的进化树,为什么还要提取?如果是读取, 用ape程序包的read.tree 函数即可。
③使用R中的FD、picante程序包具体进行NRI和Rao二次熵两个参数的计算?
picante程序包不能用来计算NRI。我没有用过FD程序包, 所以并不清楚如何用它计算Rao。在picante程序包中,Rao’s Quadratic Entropy 可以通过函数 raoD 计算,示例如下:
1 data(Phylocom)raoD(Phylocom$sample)raoD(Phylocom$sample, Phylocom$phylo)
提问 :
我想用Rao二次熵功能多样性指数分析一下群落构建,请问计算Rao coefficient的函数用哪个,dbFD ()?还是divc()?。如果要进行随机抽样计算预期值,您有没有现成的R脚本可供参考?
回复 :
基于功能性状举例矩阵的 Rao’s Quadratic Entropy, 在计算时, 用FD程序包提供的dbFD即可 (常基于gower距离)。 divc是ade4程序包下的函数, 也可以计算Rao’s Quadratic Entropy, 但是计算时有些麻烦, 参数整理不好, 容易出错。
FD程序包的结果, 应该是可信的。
基于随机抽样, 类似也有几种零模型,这和Phylocom软件中介绍的群落系统发育的零模型应该是非常类似的:
物种的距离随机化
物种在群落中随机排列
物种从一个物种库中随机抽取
independen swap (关于该模型的意义, 本人保持怀疑)
下面这个函数, 可以用来将性状矩阵的物种名, 随机重排, 生成一次null community, 将该过程重复多次,每次都计算Rao’ D, 即可得到 Rao’s D的零分布。
1 library(FD)dummy$abunnull.community <- function(x){ x.name <- rownames(x) x.name2 <- sample(x.name) rownames(x) <- x.name2 return(x)}
以上是个人理解, 与您探讨。
进化树上的孤立节点Singletons 提问 :
我用Phylocom软件创立工作路径时这样是对的吗?然后我用bladj为进化树拟合枝长时得到之后用Figtree打开这个文件时没看见进化树,是怎么回事啊?谢谢
这个sample文件里面的内容具体怎么编排呢,能否各个模板?后面的comstruct,pd,comdist的计算跟NRI和NRT的计算有什么具体的关系呢?看了你的博客和Phylocom软件使用说明了,还是一头雾水。麻烦你了。。。谢谢
回复 :
这是因为Phylocom BLADJ生成的进化树中含有singletons, 也就是进化树中某一个节点下面, 只有一个节点。
参见Phylocom 的例子, Phylocom-4.1\example_data ,里面有详细的说明。
为什么R不能读取Phylomatic生成的进化树? 提问 :
我在用R读用Phylocom拟好的带枝长的进化树out.tre的时候出现错误如下:
1 > tr <- read.tree("out.tre")错误于read.tree("out.tre") : The tree has apparently singleton node(s): cannot read tree file.Reading Newick file aborted at tree no. 1
在网上没有查到对应的解决办法,于是尝试去掉最外层括号和euphyllophyte,仍然提示相同错误。后来我用在线Phylomatic和davis带枝长的newick文件直接拟合出带枝长的进化树Phylomatic_newick.txt,改文件名后缀为.tre,用R读取时又提示相同错误:
1 >tr <- read.tree("Phylomatic_newick.tre")错误于read.tree("Phylomatic_newick.tre") : The tree has apparently singleton node(s): cannot read tree file.Reading Newick file aborted at tree no. 1
我现在不知道该怎么办了。你能帮我看下是什么问题么?
回复 :
ape程序包是不能读取含有singleton节点的进化树的。一般来说,进化树内的任何一个节点,都应该至少有两个分支。而 Phylomatic软件生成的进化树,如果在建树时没有选择clean,就会出现在很多一个节点下, 只有一个子节点的情况。 带有singletons的newick进化树,很多软件都是不能处理的,常见的包括ape, FigTree, MEGA等。
Phylomatic建树 Singletons问题 提问 :
我在用Phylomatic建树的操作过程中遇到2个问题,向您请教:
能生成没枝长的系统树,并能在treeview中读取;
生成的有枝长的系统发育树out.tre文件在treeview中打不开,提醒是file is not recognised tree file或者extra pair of parentheses;
附件是我在操作过程中用的物种信息及相关的文档,请指教。
回复 :
由于Phylocom BLADJ算法在计算中, 引入了很多节点, 而每个节点下只有一个分支,就构成了所谓的Singletons。 而在进化树的定义中, 只有一个节点的分支, 是不允许的, 很多软件是不支持这样的进化树的。 例如ape, Figtree等, 都不支持拥有singleton的进化树。
1.能生成没枝长的系统树,并能在treeview中读取;
基于物种名录,Phylomatic生成的进化树,选择clean,是没有singleton的, 故可以读取, 并且绘图。
2.生成的有枝长的系统发育树out.tre文件在treeview中打不开,提醒是file is not recognised tree file或者extra pair of parentheses;
经过BLADJ拟合枝长之后, 生成的进化树, 因为含有Singletons, 所以不能读取。目前只知道Dendroscope可以绘制含有Singletons的进化树。
R软件在群落系统发育分析中的应用 学习R的入门资料 提问 :
对于学习R,有些什么文献或资料可以推荐的吗?
回复 :
建議參考 E. Paradis的 Analysis of Phylogenetics and Evolution with R
R 软件怎样为每个样方建立进化树? 提问 :
假设我进行了100个植物样方的调查,共记录植物物种500种,根据调查记录物种建立了500个种的谱系树,怎样根据这个总的谱系树来建立每个样方的谱系树(subtree)呢?应该用R的什么包?
回复 :
如果真是需要建立每个样方的谱系树, 则用ape程序包的drop.tip 函数, 从总体的进化树中, 去除掉该样方中没有出现的种即可。
提问 :
但这样的话就需要每个样方单独做。是否还有批量处理的方法呢?
回复 :
用for 循环, 每个小样方单独做。
怎样计算物种之间的系统发育距离? 提问 :
想请教您一个有关植物谱系树的问题。我们有三个物种(木荷、小叶青冈、栲树),想知道其谱系距离,所以用Phylocom软件的bladj来弄含枝长的谱系树,便能把phylo文件中的内部节点和年龄整合到谱系树中,我想问一下这里的ages文件中三个物种的年龄怎么来确定呢?(我用软件自带的ages文件里定义的年龄,并没有找到我需要的物种),感谢帮助。
回复 :
按: 提问人并不需要知道ages文件是怎么来的, 他想了解的是如何计算物种之间的系统发育距离。
Phylocom 和 Phylomatic建立的进化树是不能用来估计同一个科内部的物种系统发育距离的。 这是因为其给出的年龄为科的起源时间,而科下各属、种均没有足够的时间信息, 所以Phylocom的BLADJ算法自动将科以下的时间三等分。 这种算法在进化树覆盖很多距离很远的分类单元时可以近似认为是合理的。 但是在物种很少,且系统发育关系很近时则是非常不合理的。
你提到的三个种分科如下:木荷(Schima superba , Theaceae)、小叶青冈(Cyclobalanopsis myrsinaefoli a, Fagaceae)、栲树(Castanopsis fargesii , Fagaceae)已经包含在 David C. Tank等人的进化树中。 该树的枝长为分化时间。可以用R获取。
附件中是相应的R脚本, 数据文件和结果。供参考。
1 library(ape)tree <- read.tree("Vascular_Plants_rooted.dated.tre")#### https://github.com/ropensci/rgbif_mcglinn/blob/master/PhylogeneticResources/Vascular_Plants_rooted.dated.tretips <- tree$tip.labeltips.to.delete <- tips[!tips %in% c("Schima_superba", "Quercus_myrsinifolia", "Castanopsis_fargesii")]tree2 <- drop.tip(tree, tips.to.delete)plot(tree2)taxa.dist <- cophenetic(tree2)write.csv(taxa.dist, "taxa.distance.csv")write.tree(tree2, "tree2.tre")
根据已有的大型进化树,能够获得只有部分分类单元的进化树? 提问 :
比较大的系统发育树里能否挑选出部分物种,重新构建系统发育树呢?新构建的系统发育树能否通过Phylocom软件计算其多样性等相关变量呢?
回复 :
如果当前的进化树是有枝长的newick格式的进化树(一定要保证枝长表示的是分化时间), 要选出部分的种类,可以先用R软件ape程序包的read.tree读取进化树, 之后用drop.tip() 函数去掉不需要的物种。
新的系统发育树可以用Phylocom软件计算PD, comstruct, phylobeta等
R怎样调用Phylocom? 提问 :
想问下R调用Phylocom要大概如何做?sandel的包是不是PhyloMeasures包?但是我在文件夹中test.Phylocom.R文件中,不知道具体求betaMNTD和MNTD函数格式是什么?
我在里面原始文件是不是用
1 source("Phylocom.R")par(ask=TRUE)a <- readsample("sample")library(picante)p <- read.tree("pruned.tre0")#或者用 /p <- readphylo("pruned.tre0")
回复 :
可以直接使用
1 system("Phylocom pd -s sample3 -f pruned.tre0 -m 0 -r 9999 -a>pd.output.txt")system("Phylocom pd -s sample3 -f pruned.tre0 >pd.txt") system("Phylocom comstruct –s sample3 -f pruned.tre0 -m 0 -r 9999 -a>mpd.output.txt")system("Phylocom comstruct –s sample3 -f pruned.tre0 -m 0 -r 9999 -a -v>mpd.output.txt") system("Phylocom comdist –s sample3 -f pruned.tre0 -m 0 -r 9999 -a -n>ses.betampd.output.txt")system("Phylocom comdistnt –s sample3 -f pruned.tre0 -m 0 -r 9999 -a -n>ses.betamntd.output.txt")
但是如果直接运行Phylocom的内存不够, 在R中调用Phylocom仍然会内存不足的问题。 如果用picante或者PhyloMeasures程序包,请一定要按照函数的说明准备输入的数据。
怎样将进化树导入R?怎样使用Phylocom? 提问 :
我将树以newick的形式导进去后无法进行后面的分析,我也不清楚像样方数据怎么输到R中。说明文件中的例子我操作了一次,也成功了,但Phylocom是如何输入进去的我不清楚。您能否教我使用这个程序包,举一个三、五个样方的例子?
回复 :
read.tree读取多棵进化树 提问 :
如何一次性地读入多个.tre或.nex格式的文件到R的一个list中?
回复 :
ape的read.tree返回值是list,可以读取多棵进化树,可参考如下命令:
1 read.tree(file = "", text = NULL, tree.names = NULL, skip = 0, comment.char = "#", keep.multi = FALSE, ...)
R中的距离矩阵怎样转换为data.frame? 提问 :
我这边有两个100×100的对角矩阵,一个是betaNTI值,同时我想和样点间pH的差值比较,dist(data$ph),以一个散点图的形式呈现,但是我不知如何通过R把他们的所有距离一一对应起来?我对循环不熟df <- data.frame()不知道如何能把这个distz转换成一列向量啊?大概应该有5050值吧?
1 for (i in 1:99){ t <- ntinew[((i+1):100),i]; df <- rbind(t[i])}df
回复 : 可以先将对角线的值设为0,然后转换成dist类型的距离矩阵,最后用simba程序包的liste函数,将其转换为data.frame即可。
picante 运行picante下标出界 提问 :
我建立了一个树Phylo2.txt;同时有一个群落data2.txt。我计算能得到PD,但是我计算MPD或者MNTD时,却显示是下标出界,我好久都找不出原因。我现在把所有的东西都简化了,还是找不出原因,不知您能不能给帮忙解答一下!
回复 :
下标出界很可能是进化树中的物种数与群落中的物种数不匹配造成的。需要将两者的名称完全对应起来,不能一个多于另一个。用Phylocom计算就可以发现这个问题了。
Phylocom的NRI和picante包的ses.mpd()哪个更准确?怎样用Phylomatic? 提问 :
我用Phylocom的数据进行练习,发现Phylocom软件计算的NRI值跟我用picante包ses.mpd()计算的值,差了一个负号,而且两者的值也不太一样,是不是因为Phylocom计算结果比用picante包的结果更精确呢?有这样的说法吗?
在Phylomatic 中,无论有没有勾选clean,按照练习的步骤在Phylocom 中进行练习,然后并没有得到结果,我不知道我是哪里出错了。
回复 :
Phylocom计算的是NRI, picante计算的是SES.mpd,两者符号相反。picante和Phylocom的结果都是可以使用的,在精确程度上, 并没有什么明显区别。
结果绝对值的差异主要是来源于随机化过程,在R中, 可以设定随机数种子, 保证每次获得的随机数序列都是相同的。 但是在Phylocom中似乎不能设定,因此, Phylocom每次运行的结果会有一定的差异。
第二个问题,很可能是你的命令设定错了。 这跟是否勾选clean没有什么关系。 也可能是你的sample的格式有问题, 或者sample中的物种名与进化树中的物种名不能完全匹配。具体要看Phylocom在运行时给出什么提示。
picante中sample和tree的格式 提问 :
我最近在学习R的程序包picante分析PSV。我用lm.str()打开R自带的sample和tree以后,仍然不太明白可以用R进行分析的sample和tree的具体格式。我原来的sample格式都是对应Phylocom软件建的。
回复 :
sample 的格式, 参见
1 data(Phylocom)Phylocom$samplePhylocom$phylo
sample可以由 picante程序包的 sample2matrix() 函数户获得, phylo可以由 ape程序包的 read.tree() 获得。
picante计算PD以及SES.PD 提问 :
我尝试着用R软件使用您的代码分析我的野外调查数据时,当运行到代码
1 pd_red <- pd(samp = mat, tree = tree, include.root=TRUE)
时,发生以下错误:
1 “Error in phy$edge[sndcol, 2] <- newNb[phy$edge[sndcol, 2]] <- (n + 2):(n +: number of items to replace is not a multiple of replacement lengthIn addition: Warning message:In newNb[phy$edge[sndcol, 2]] <- (n + 2):(n + phy$Nnode) :number of items to replace is not a multiple of replacement length”
不知道您在使用过是是否有这样的情况,一般是什么原因引起这样的错误呢,我应该怎么排除啊。
回复 :
我的电脑上运行没有问题。 我的是 64bit, Win10. 脚本和结果参见附件。
1 setwd("C:\\Users\\jlzhang\\Desktop\\experiment0310")library(picante)dat <- read.table("sample", header = FALSE)mat <- sample2matrix(dat)tree <- read.tree("phylo")#### Compare the tip labels wpd_red <- pd(samp = mat, tree = tree, include.root=TRUE)ses.mpd(samp = mat, dis = cophenetic(tree), null.model="taxa.labels" )
为什么picante的ses.mpd和Phylocom的NRI符号相反? 提问 :
我随机选的物种和随机放置不同的样方中所进行的练习,用picante包计算它们的谱系结构。然后软件求得的RNI值,跟我用公式计算的值数值是一样的,但是公式求得值是负数呀,我想问下这两个结果都是一样的吗?
回复 :
picante包计算出来的是ses.mpd或ses.mntd。Phylocom计算的是NRI或NTI。两者符号相反。
应该用NRI还是SES.mpd? 提问 :
如果根据picante包计算的结果,代入这个公式的话,结果为负值-0.3936901,图中的mpd.obs.z是正值,picante包里的说明是这个mpd.obs.z相当于NRI值,这样的话最后我是取哪个值呢?
回复 :
取哪个值都可以。 这两个指数只相差一个负号。 引用正确的参考文献就可以了。 如果是用Phylocom计算的, 还是用NRI,如果是用picante计算的,建议用SES.mpd